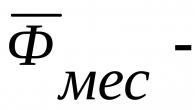L'atrophie musculaire spinale est une pathologie grave qui implique une altération de la fonction motrice. Il existe quatre types de maladie, parmi lesquels l'amyotrophie spinale de Werdnig-Hoffmann, qui se développe pendant la petite enfance et l'enfance, est considérée comme la plus défavorable. Ce type de pathologie est héréditaire et incurable, et les techniques utilisées ne peuvent que légèrement améliorer l’état du patient. Par quels signes la maladie est-elle reconnue, et que faire si elle est détectée ?
L'amyotrophie musculaire spinale, ou SMA, implique des dommages aux neurones moelle épinière responsable de fonctions motrices muscles. Ce sont généralement les muscles des jambes et du cou qui souffrent le plus, mais les muscles membres supérieurs sont moins fréquemment touchés. Les patients ont des problèmes de mouvement, de déglutition, de tenue de la tête, mais la sensibilité est préservée et il n'y a aucun retard dans développement mental. Mais si avec d'autres formes de SMA, les patients ont une chance de vivre jusqu'à un âge avancé, bien qu'avec un handicap, alors avec l'amyotrophie de Werdnig-Hoffmann durée maximale la vie ne dépasse pas 30 ans.

La pathologie est assez rare - dans un cas sur 80 à 100 000. Mais il existe bien plus de porteurs du gène responsable du développement de l’anomalie. La maladie est héritée de manière autosomique récessive et pour qu'un enfant présente une amyotrophie de Werdnig-Hoffmann, les deux parents doivent être porteurs du gène. Bien que dans ce cas, la probabilité de maladie chez le bébé ne soit que de 25 %. La cause de la maladie est uniquement une prédisposition génétique ; aucun lien entre la SMA et les blessures, infections et autres facteurs n'a été trouvé.

Manifestations de la maladie
Les experts distinguent trois formes de SMA de Werdnig-Hoffmann, qui diffèrent en termes de manifestation et de symptômes typiques.
Tableau. Formulaires Werdnig-Hoffmann SMA
Toutes ces formes de pathologie sont unies par l'absence de toute déficience mentale et sensorielle, mais le tableau clinique présente des différences significatives.

Forme infantile
Dans la pathologie de type I, les premiers symptômes sont perceptibles dès la naissance d'un enfant : il naît avec une parésie flasque, produit des cris très faibles et il n'y a pas de réflexes profonds. Il n'est pas difficile pour un spécialiste de déterminer l'hypotonie musculaire, ce qui permet d'établir la présence d'une atrophie de Werdnig-Hoffmann dès les premiers jours. Ces enfants tètent lentement le sein, avalent mal le lait et s'étouffent souvent. Même les mouvements de la langue sont difficiles et, après un examen attentif, on peut y voir des contractions musculaires involontaires - de petits mouvements ondulatoires. Tout cela entraîne des difficultés d'alimentation, car la nourriture peut pénétrer dans Voies aériennes et provoquer la mort de l'enfant.

En plus de ces symptômes, on observe une parésie du diaphragme et une déformation du squelette : l'enfant peut avoir une colonne vertébrale courbée, déprimée ou, à l'inverse, fortement saillante. cage thoracique, les joints sont démoulés. Les enfants avec ce diagnostic ont un développement moteur nettement en retard par rapport à leurs pairs. Ils ne peuvent pas tenir la tête haute, se retourner d’un côté à l’autre, atteindre un objet qui attire l’attention ou prendre une position assise. En même temps, les expressions faciales et les fonctions muscles des yeux la maladie n'affecte pas et les émotions exprimées par l'enfant ne sont pas déformées.
Pour information : chez un nombre limité d'enfants atteints d'amyotrophie de Werdnig-Hoffmann, les capacités motrices apparaissent encore, quoique avec beaucoup de retard, mais ensuite après un bref délais régresser.
La SMA de type 1 s'accompagne souvent d'autres pathologies congénitales :
- dysplasie des articulations pelviennes;
- hydrocéphalie du cerveau;
- les hémangiomes;
- malformation cardiaque.

La maladie se développe très rapidement et la mort survient dans la plupart des cas dans les six mois suivant la naissance. Certains bébés vivent jusqu'à 2-3 ans. La cause du décès est généralement une forme grave de maladie cardiaque et arrêt respiratoire.
Forme précoce
Avec le deuxième type d’amyotrophie, les enfants se développent tout à fait normalement au cours des six premiers mois de leur vie, sans aucun signe avant-coureur. À six mois, certains commencent même à se lever assez activement et à se déplacer le long du berceau ou du parc. Le premier symptôme est une faiblesse musculaire, qui peut se développer progressivement ou survenir soudainement dans le contexte de tout maladie infantile, diverses infections. Cela se produit généralement entre 7 et 10 mois de la vie du bébé.

Les fonctions musculaires sont d'abord perturbées dans les membres inférieurs, ce qui fait que l'enfant rampe de plus en plus mal et a des difficultés à se tenir debout. De plus, la lésion s'étend de plus en plus haut, ce qui entraîne une diminution des réflexes profonds, l'apparition de tremblements des doigts, des contractions musculaires volontaires de la langue et un dysfonctionnement du système respiratoire. Cette forme de pathologie survient moins intensément que dans l'enfance et la plupart des patients survivent jusqu'à l'adolescence, même si la qualité de vie reste très faible pendant cette période. Ces enfants ne peuvent pas prendre soin d’eux-mêmes et ne peuvent se passer d’une aide extérieure. La cause du décès est généralement un dysfonctionnement organes respiratoires, et diverses infections, qui touchent souvent un organisme fragilisé par la maladie.
Forme tardive
Les experts considèrent cette forme de SMA comme la moins grave, même si une personne peut vivre avec elle pendant 30 ans maximum. Manifestations typiques surviennent le plus souvent lorsque l'enfant a entre 1,5 et 2 ans. Jusqu'à ce moment là Développement physique se passe sans la moindre infraction Le bébé marche, court et présente des niveaux d'activité normaux pour son âge. Seul un nombre limité de patients peuvent présenter des retards de développement moteur ou une lenteur excessive.

Les premiers symptômes sont légers :
- l'enfant se fatigue plus vite ;
- la coordination des mouvements diminue, le bébé tombe plus souvent en courant et en marchant ;
- une léthargie est souvent observée.
Au fur et à mesure de l’évolution de la maladie, d’autres signes plus caractéristiques apparaissent :
- la démarche change, l'enfant lève les genoux plus haut en marchant ;
- la faiblesse musculaire augmente;
- observé léger tremblement des doigts;
- il y a des spasmes volontaires de la langue, des difficultés avec les fonctions de déglutition ;
- Des déformations des os et des articulations se développent, elles sont particulièrement prononcées au niveau de la poitrine.

Tous ces processus se développent assez lentement et la capacité de marcher persiste jusqu'à l'âge de 8 à 10 ans environ. À l’avenir, les déplacements ne seront possibles qu’en fauteuil roulant, mais la capacité de prendre soin de soi ne sera pas complètement perdue avant plusieurs années. Avec une thérapie de soutien, les personnes présentant ce diagnostic vivent jusqu'à 25 à 30 ans.
Diagnostic de la maladie
Le syndrome d'hypotonie musculaire congénitale est caractéristique non seulement de l'AMS, mais également d'un certain nombre d'autres pathologies, par exemple la paralysie cérébrale, sclérose amyotrophique, myopathies différentes formes. Par conséquent, un examen visuel, ainsi que des informations sur le moment de l'apparition des symptômes et la dynamique de leur développement, ne permettront pas de différencier l'amyotrophie de Werdnig-Hoffmann. La tomodensitométrie ou l'IRM peuvent exclure la présence d'autres pathologies de la colonne vertébrale, mais ces études ne suffisent pas à déterminer la SMA.

Principal méthode de diagnostic si une atrophie musculaire spinale est suspectée, une électroneuromyographie, ou ENMG, est réalisée. Cette méthode conçu pour analyser le fonctionnement du système neuromusculaire, en particulier le passage de l'influx nerveux et la réaction à celui-ci. Pour confirmer le diagnostic, une biopsie du tissu musculaire et des tests ADN sont également prescrits.
Important! Il est conseillé aux personnes ayant des antécédents familiaux de SMA de se soumettre à des tests génétiques pour déterminer la présence du gène SMN, responsable de troubles du développement musculaire. Les femmes enceintes subissent un test ADN prénatal du fœtus, et si le diagnostic est confirmé, il s'agit d'une indication sérieuse d'interruption de grossesse.
Thérapie d'entretien
La SMA de Werdnig-Hoffmann est incurable, c'est pourquoi les patients bénéficient d'un traitement de soutien visant à atténuer les symptômes de la maladie. L'accent principal est mis sur l'amélioration du métabolisme des fibres musculaires et nerveuses affectées, ce qui contribue à ralentir la progression de la pathologie. A cet effet, ils utilisent médicaments plusieurs groupes : neurométabolites, médicaments pour faciliter la transmission neuromusculaire, médicaments pour améliorer le trophisme tissulaire et la circulation sanguine.
Le plus souvent prescrit :

Le type de médicaments, la posologie et la durée d'utilisation sont déterminés par le médecin traitant en fonction des résultats de l'examen et en tenant compte de l'âge de l'enfant. En plus des médicaments, il est indiqué physiothérapie, procédures physiothérapeutiques, massage doux. En cas de déformation grave de la colonne vertébrale, utilisez corsets orthopédiques et des bandages.
Vidéo - Amyotrophie spinale de Werdnig-Hoffmann
L'atrophie musculaire n'est pas la seule pathologie système moteur, qui est transmis génétiquement. Vous pouvez découvrir quelles autres maladies héréditaires de la colonne vertébrale existent et comment y faire face.
L'atrophie musculaire spinale (AMS), ou amyotrophie spinale de Werdnig-Hoffmann, est une maladie héréditaire autosomique récessive caractérisée par une hypotension progressive et une faiblesse musculaire.
L'affaiblissement caractéristique du tissu musculaire est dû à la dégénérescence progressive des motoneurones alpha de la corne antérieure de la moelle épinière. Ainsi, la maladie repose sur une pathologie de la moelle épinière, qui peut être héréditaire.
Une caractéristique de la maladie est une manifestation plus active de faiblesse les muscles squelettiques situés plus profondément que ceux situés plus près de la surface du corps. Dans ce document, nous parlerons des symptômes et du traitement de l'amyotrophie spinale de Werdnig-Hoffmann.
Ces informations seront utiles à tous ceux qui, pour diverses raisons, ont dû faire face à cette maladie grave et souvent mortelle.
Chez certains patients de processus pathologique les motoneurones des nerfs crâniens, notamment V à V, peuvent également être impliqués XIIe paire. Dans ce cas, la maladie provient de corne postérieure les cellules de la moelle épinière, qui en plus de tout provoquent une insuffisance des muscles du diaphragme, tube digestif, cœur et sphincters.
En 1890, Werdnig a décrit pour la première fois la forme infantile classique de SMA, une manifestation du syndrome chez les jeunes enfants. Plusieurs années plus tard, en 1956, Kugelberg et Welander classèrent moins forme grave atrophie musculaire spinale chez les patients âgés.
Grâce à ces scientifiques, les médecins peuvent aujourd'hui différencier avec précision la SMA de différents types de maladies présentant des symptômes similaires, par exemple la dystrophie musculaire de Duchenne.
L'amyotrophie spinale est le diagnostic le plus courant chez les filles, avec une faiblesse sévèrement progressive. C'est l'un des plus courants raisons génétiques mort chez les enfants.
Le syndrome SMA est divisé en quatre types en fonction de l’âge du patient :
- Type I (amyotrophie spinale de Werdnig-Hoffmann). Se développe jusqu'à l'âge de 6 mois.
- Type II – entre 6 et 12 mois.
- Type III (maladie de Kugelberg-Welander) – âgés de 2 à 15 ans
- Type IV chez les patients adultes.

Diffusion
L'incidence de la maladie est d'environ un cas pour 15 à 20 000 personnes. Si nous parlons uniquement des nouveau-nés, ce chiffre sera d'environ 5 à 7 cas pour 100 000. L’amyotrophie spinale étant une maladie héréditaire récessive, de nombreux parents peuvent en être porteurs sans le savoir.
La prévalence des porteurs de SMA est de 1 sur 80 ; en d’autres termes, une famille sur 80 peut avoir un enfant souffrant d’amyotrophie spinale. Ce risque augmente plusieurs fois lorsque les deux parents sont porteurs du gène mutant.
Ainsi, le syndrome SMA est la maladie dégénérative du système nerveux la plus fréquente chez les enfants après la mucoviscidose et, comme indiqué ci-dessus, c'est la principale pathologie. cause héréditaire mortalité infantile.
Le décès est dû à une insuffisance respiratoire. Comment patient plus jeune sur étapes préliminaires maladie, plus le pronostic est mauvais. Général âge moyen au moment du décès, il y a environ 10 ans. L’état d’intelligence et d’autres indicateurs du développement psychosomatique de l’enfant n’affectent en rien la progression de la maladie.
Contrairement aux patients jeunes, les hommes adultes sont plus souvent touchés par la maladie que les femmes dans un rapport d'environ 2 : 1, tandis que l'évolution clinique chez les patients de sexe masculin est plus sévère. L’augmentation des cas de maladie chez les femmes commence vers l’âge de 8 ans et les garçons « rattrapent » les filles vers 13 ans.
Amyotrophie spinale - symptômes
Le premier type d’amyotrophie spinale provoque l’apparition des premiers symptômes avant la naissance du bébé. La plupart des mères signalent une inactivité fœtale anormale étapes tardives grossesse. Les symptômes de la SMA chez les nouveau-nés sont assez évidents : l'enfant ne peut pas se retourner tout seul, puis prendre une position assise.
De plus, une détérioration clinique progressive se développe, qui dans la grande majorité des cas aboutit à la mort. La mort survient généralement par insuffisance respiratoire et ses complications chez les patients âgés de 2 ans.
Les patients atteints de SMA de type 2 se développent normalement au cours des 4 à 6 premiers mois de leur vie. Ils pourront peut-être s’asseoir seuls, mais ils ne pourront jamais marcher et auront besoin d’un fauteuil roulant pour se déplacer à l’avenir. En règle générale, ces enfants vivent beaucoup plus longtemps que les patients souffrant de atrophie spinale Werdnig-Hoffmann. La durée de vie moyenne peut atteindre 40 ans.

Les patients atteints de type 3 ont souvent des difficultés à monter les escaliers ou à se lever du sol, principalement en raison d'une faiblesse des extenseurs. articulation de la hanche. L'espérance de vie est proche de la normale.
En savoir plus sur les symptômes de la SMA
Les nouveau-nés atteints d’amyotrophie spinale de type 1 sont inactifs. Ils bougent leurs membres avec beaucoup de difficulté, voire pas du tout. Les hanches sont presque constamment pliées, affaiblies et peuvent facilement être tordues à la main dans différentes directions. Les genoux sont également fléchis.
Comme les muscles externes sont généralement moins touchés, les doigts et les orteils bougent presque normalement. Les bébés ne peuvent pas contrôler ou lever la tête. L'aréflexie (manque de réflexes) est observée chez presque tous les patients.
Les enfants souffrant du deuxième type de SMA sont capables de bouger la tête et 75 % de ces patients peuvent s'asseoir de manière autonome. La faiblesse musculaire est plus importante dans les membres inférieurs que dans les membres supérieurs. Réflexe rotule absent. Les enfants plus âgés peuvent démontrer des réflexes biceps et triceps.
La scoliose est la plus symptôme courant SMA, et la plupart des patients développent une luxation de la hanche, unilatérale ou bilatérale. Ces signes apparaissent avant l'âge de 10 ans.
Les patients atteints d’amyotrophie spinale de type 3 peuvent marcher tôt dans la vie, et cette capacité peut être maintenue en ambulatoire tout au long de l’adolescence. La faiblesse peut entraîner une endurance limitée du corps. Un tiers des patients se retrouvent en fauteuil roulant à 40 ans.

Traitement
Il n'y a actuellement aucun connu traitement médical atrophie musculaire spinale, il convient donc de noter immédiatement que le taux de survie chez les patients d'âge précoce et moyen est assez faible.
Les nouveau-nés atteints d'amyotrophie spinale de Werdnig-Hoffmann nécessitent peu de soins orthopédiques en raison de leur courte espérance de vie. L'attelle est utilisée dans les cas souvent observés avec une activité musculaire affaiblie.
Pour les patients atteints de SMA de type II et III, thérapie physique peut être utilisé pour traiter les contractures articulaires (mouvements limités). Pour plus traitement radical Un traitement chirurgical est indiqué pour les contractures.
Comme indiqué ci-dessus, le problème orthopédique le plus courant dans le syndrome SMA est la scoliose, qui prend souvent une forme grave. La progression de la courbure rachidienne est d'environ 8° par an, malgré le traitement par appareil orthodontique.
L’arthrodèse vertébrale postérieure de type segmentaire est souvent recommandée chez les jeunes patients présentant une courbure colonne vertébrale ne peut pas être corrigée par un appareil orthodontique, ainsi que chez les patients de plus de 10 ans présentant une courbure supérieure à 40°.
La chirurgie doit être retardée jusqu'à ce que point médical c'est possible. Il convient de garder à l'esprit que la progression de la courbure se produit plus lentement chez les patients atteints du troisième type de SMA et apparaît plus souvent chez les patients plus âgés. âge tardif.

Régime
Le traitement de l’amyotrophie spinale nécessite une approche individuelle du rationnement du menu du patient, qui, malheureusement, n’est très souvent pas respectée par les médecins traitants. Les indicateurs anthropométriques, la composition sanguine et les marqueurs biochimiques de l'état musculaire sont éléments importantsévaluations chez les patients atteints de SMA.
La particularité de l'évolution de la maladie chez un patient particulier peut nécessiter une intervention dans son alimentation afin d'influencer les indicateurs ci-dessus, puisque c'est à l'aide de la nourriture que les muscles peuvent recevoir ces nutriments dont le patient a besoin dans son cas.
Bien sûr, cette approche du traitement amyotrophie musculaire spinale n'est pertinent que lors du diagnostic de la deuxième ou de la troisième forme de la maladie.
Thérapie physique
Un soutien supplémentaire pour un patient souffrant d'amyotrophie spinale des deuxième et troisième types n'est pas moins important que l'alimentation. Tout d'abord, avec l'aide d'une charge normalisée, vous pouvez empêcher la progression de la contracture articulaire, ainsi que maintenir la force, l'endurance et l'indépendance dans les soins personnels.
L'exercice physique joue un rôle assez important dans les activités éducatives, sociales, psychologiques et professionnelles du patient, puisqu'il aura la possibilité de mener une vie presque normale, comme les personnes en bonne santé.
Il s'agit d'un groupe de maladies héréditaires dont la principale caractéristique est des lésions des motoneurones des cornes antérieures de la moelle épinière, ainsi que des lésions des racines des nerfs crâniens IX, X, XII.
L'amyotrophie spinale est caractérisée par une violation de l'innervation musculaire des membres inférieurs, cou, tête, muscles respiratoires. Critères importants pour la définition bon diagnostic c’est la préservation de tous les types de sensibilité, le développement normal des muscles des membres supérieurs et l’absence de l’enfant les troubles mentaux. L'incidence de la maladie est de 7 personnes pour 100 000 naissances.
Causes de l'amyotrophie de Werdnig-Hoffmann
Le gène de l'amyotrophie spinale de Werdnig-Hoffmann (SMN) est localisé sur le chromosome V et est hérité de manière autosomique récessive. Les parents dont les chromosomes portent le gène SMN ont 25 % de chances de produire un enfant atteint d'amyotrophie spinale.
Modifications pathomorphologiques de l'amyotrophie de Werdnig-Hoffmann
Au cours de l'étude, une diminution du volume de la moelle épinière est constatée. Les cellules ganglionnaires s'atrophient ou disparaissent complètement. Dans les racines antérieures, une dégénérescence, une démyélinisation, des modifications sclérotiques des fibres nerveuses (péri-, épi-, endoneuriales) avec dépôt de graisse sont détectées. Dans les muscles squelettiques, on constate des faisceaux atrophiés entrelacés de fibres intactes et une prolifération. tissu conjonctif.
Classification de la maladie amyotrophie de Werdnig-Hoffmann
Par moment d'occurrence et degré changements dystrophiques pour l'amyotrophie spinale Werdnig-Hoffmann :
- Congénital (apparition des symptômes de la maladie au cours des 6 premiers mois de la vie) ;
- Petite enfance (de 6 mois à 1 an et demi) ;
- Fin de l'enfance (plus de 1,5 ans).
Symptômes de l'amyotrophie de Werdnig-Hoffmann
Le plus lourd est forme congénitale Amyotrophie spinale de Werdnig-Hoffmann. Chez les enfants, dès les premières minutes de la vie, parésie flasque. Une faiblesse musculaire, une diminution des réflexes de la période néonatale ou leur absence sont révélés. Les nouveau-nés tètent faiblement le sein, ils ont des contractions fasciculaires de la langue et la déglutition est difficile.
Cette forme de la maladie s'accompagne de la formation de déformations musculo-squelettiques, notamment scoliotiques ; poitrine en forme d'entonnoir ou de « poulet » ; contractures articulaires. Très Dans certains cas l'enfant a la capacité de tenir la tête haute et de s'asseoir. Cependant, ces capacités se développent tardivement puis régressent. Cette maladie s'accompagne souvent anomalies congénitales, comme l'hydrocéphalie, la dysplasie des articulations de la hanche, les déformations planovalgus ou planovarus des pieds, les testicules non descendus dans le scrotum, les hémangiomes, etc. Les enfants meurent avant 9 mois (moins souvent jusqu'à 2 ans) d'une insuffisance cardiovasculaire ou respiratoire, cause de qui – hypotension muscles pectoraux et les muscles du diaphragme.

Formes de la petite enfanceet l'amyotrophie spinale de Werdnig-Hoffmann se caractérise par une manifestation dans la seconde moitié de l'année. Un enfant malade commence rapidement à relever la tête, à s'asseoir et parfois même à se tenir debout ou à marcher. De plus, après avoir souffert d'une entérocolite alimentaire, la maladie progresse : une parésie flasque apparaît d'abord sur les jambes, puis s'élève vers le corps et les membres supérieurs. En raison d'une atrophie musculaire diffuse, on note des contractions fasciculaires de la langue, des contractures et de fins tremblements des mains. Syndrome bulbaire se développe beaucoup plus tard. La forme de la petite enfance de l'amyotrophie spinale de Werdnig-Hoffmann n'est pas aussi maligne que la première variante de la maladie, cependant, la mort survient entre 12 et 15 ans.
La forme tardive de la maladie se manifeste chez les enfants d'âge préscolaire. Sur fond de bien-être imaginaire, lorsque l'enfant bouge de manière autonome, saute, court, des raideurs apparaissent, les mouvements deviennent maladroits (démarche d'une « poupée à remonter »), les enfants trébuchent souvent. L'atrophie des muscles squelettiques se produit progressivement et lentement : d'abord, une parésie flasque est observée dans les parties inférieures des membres inférieurs, puis les muscles sont impliqués dans le processus. parties inférieures membres supérieurs, torse. L'atrophie musculaire peut passer inaperçue, car à cet âge le tissu sous-cutané est bien développé tissu adipeux. Progressivement, les réflexes tels que les réflexes pharyngés et palatins s'affaiblissent et les réflexes inconditionnés diminuent. La maladie s'accompagne de déformations appareil de support, le plus souvent il s'agit de poitrine de « poulet ».
Le pronostic avec un traitement adéquat et opportun est plus favorable que celui des deux premières options. Les patients peuvent vivre jusqu'à 30 à 40 ans. Cependant, la capacité de se déplacer de manière indépendante disparaît au bout de 10 à 12 ans.
Dans la littérature, on peut parfois trouver une quatrième forme de la maladie, une forme adulte, qui se manifeste à partir de 35 ans. Il s'agit d'une forme extrêmement rare et la plus favorable de la maladie, dans laquelle l'innervation des groupes musculaires des membres inférieurs est perturbée. Chez ces patients, la capacité de bouger de manière indépendante est perdue, mais il n'y a pas de problèmes de respiration ou de déglutition. La forme adulte de l'amyotrophie n'affecte pas l'espérance de vie des patients.
Diagnostic du SDA de Werdnig-Hoffmann
Le diagnostic est confirmé sur la base image clinique (début précoce changements atrophiques, Commencer changements dégénératifs dans les groupes musculaires proximaux, hypotonie musculaire, contractions musculaires de la langue, absence de pseudohypertrophie), données ENMG (électroneuromyographie), résultats de biopsie fibre musculaire, IRM, analyse généalogique (recherche de mutations génétiques chez les parents et l'enfant). La maladie évolue rapidement.
Diagnostic différentiel Amyotrophie de Werdnig-Hoffmann
1. Avec d'autres maladies caractérisées par le « syndrome de l'enfant disquette » ;
2. Maladies génétiques métabolisme;
3. Amyotrophie d'Oppenheim (actuellement considérée par certains experts comme une variante de l'amyotrophie spinale de Werdnig-Hoffmann) ;
4. Paralysie cérébrale ;
5. Progressif dystrophies musculaires(Duchenne et Erb-Roth) ;
6. Amyotrophie de Kugelberg-Welander ;
7. Intoxication au plomb.
Traitement de l'amyotrophie de Werdnig-Hoffmann
Amyotrophie spinale de Werdnig-Hoffmann sur ce moment une maladie incurable et en constante évolution. Il n'y a que thérapie symptomatique: médicaments qui agissent sur processus métaboliques Tissu nerveux(cérébrolysine ; aminolon ; encéphabol) ; nootropiques (lucétam, nootropil); vitamines groupe B; massage et thérapie par l'exercice, régime spécial etc.
Les mutations génétiques de l'amyotrophie spinale sont associées à une diminution de la production de protéine SMN, ce qui entraîne la perte de motoneurones. La tâche numéro un de la pharmacologie moderne dans cette maladie est de trouver un médicament capable d'augmenter le niveau de protéine SMN.
La prévention Amyotrophie de Werdnig-Hoffmann
Consiste en diagnostic opportun troubles génétiques chez les parents, diagnostics ADN prénatals. Lorsqu'une pathologie est détectée chez le fœtus, la question de
C'est effrayant de découvrir que le bébé ne pourra jamais s'asseoir, se lever ou courir. C’est encore plus effrayant de voir à quel point grandissent normalement et enfant en développement commence soudainement à disparaître lentement, tombe constamment, après quelques mois, il ne peut plus monter les escaliers et un jour, il perd la capacité de simplement se lever.
Les médecins regroupent plusieurs types de maladies héréditaires caractérisées par des troubles du mouvement en un seul groupe appelé atrophie musculaire spinale. Dans la CIM-10, ils sont codés G12 avec des indications supplémentaires sur le type de maladie.
Selon les chercheurs, environ 0,01 à 0,02 % des enfants naissent avec un diagnostic de SMA. La pathologie survient plus souvent chez les garçons et les hommes.
L'amyotrophie spinale touche principalement les enfants jeune âge. Cependant, certaines formes de la maladie commencent à apparaître uniquement chez les adolescents ou les adultes. Le caractère insidieux de la pathologie réside dans le fait qu'elle enlève progressivement, jour après jour, aux patients ce qu'ils ont pu réaliser.
La pathologie a été décrite pour la première fois par G. Werdnig. Il attire l'attention sur l'atrophie équilatérale de la moelle épinière, de ses cornes antérieures et des racines nerveuses périphériques en 1891. Déjà en 1891 l'année prochaine J. Hoffman a pu prouver que nous parlons deÔ maladie indépendante. Au milieu du 20ème siècle. les chercheurs E. Kugelberg et L. Welander ont décrit une pathologie qui survient à un âge plus avancé et dont le pronostic est plus favorable.

Symptômes
Chaque type de SMA a ses propres symptômes particuliers, mais certains symptômes permettent de regrouper des maladies disparates en un seul groupe. Ce:
- Augmentation de la faiblesse musculaire et de l'atrophie.
- Avec une maladie qui apparaît après 1 à 2 ans, une dégradation des capacités déjà acquises, par exemple courir et marcher, est perceptible.
- Tremblement des doigts. Des tremblements sont également observés dans la langue.
- Déformation du squelette.
- Sécurité des personnes intellectuelles et santé mentale chez la plupart des patients.
Types de SMA
L'âge, le moment d'apparition des symptômes, les caractéristiques de l'évolution de la pathologie et le pronostic permettent de distinguer plusieurs types de maladies.
Ce formulaire la pathologie est rarement décrite ; elle est souvent associée au premier type de SMA. La maladie est congénitale. Caractérisé par absence totale mouvements, réflexes tendineux, faiblesse musculaire, mouvements limités des articulations du genou. Des troubles respiratoires ont été observés depuis la naissance.
Le diagnostic est souvent confondu avec les blessures à la naissance. Cependant, dans les deux derniers cas, les enfants s'adaptent assez vite, leur état s'améliore. Les enfants atteints de SMA ne connaissent aucune amélioration ; dans la plupart des cas, ils meurent avant d’atteindre l’âge d’un mois à cause de complications.
La pathologie du premier type a un effet très cours sévère. On l'appelle aussi maladie de Werdnig-Hoffman. Ce type peut être diagnostiqué de la naissance à 6 mois. Il existe une faiblesse musculaire et des contractions périodiques - ces dernières sont assez difficiles à voir en raison de la couche de graisse assez importante. Des tremblements peuvent périodiquement parcourir la langue du bébé.
Il y a une aggravation des nausées, des réflexes de succion, de déglutition et une altération de la salivation. Le bébé ne peut ni tousser ni crier fort. Souvent accompagné de graves troubles respiratoires, pneumonie.
La poitrine de ces enfants a une forme plus plate en raison de muscles pectoraux peu développés.
Les bébés atteints d'amyotrophie spinale de Werdnig-Hoffmann sont facilement reconnaissables à leur posture de grenouille. Hanches et épaules en abduction, coudes et genoux fléchis.
Vers 6 mois, un enfant peut apprendre à tenir la tête haute, mais ne sera presque jamais capable de s'asseoir, de se lever ou de marcher tout seul. Les problèmes de déglutition entraînent des difficultés à s’alimenter.
Cette maladie s'accompagne souvent d'oligophrénie, troubles congénitaux fonction cardiaque, petite taille de tête.
Tardive enfance
La pathologie du deuxième type se retrouve chez les enfants âgés de six mois à un an et demi à deux ans. La maladie de Dubowitz se caractérise par une faiblesse et des tremblements du sections profondes muscles, tremblements des doigts, de la langue, limitation de l'amplitude de mouvement des membres. Les enfants sont distingués peu de poids, retard de développement. Ils s'assoient et mangent eux-mêmes, mais ne peuvent ni se lever ni marcher.
La maladie est évolutive. Au fil du temps, les muscles de la poitrine et du cou s'affaiblissent, les réflexes tendineux disparaissent, des troubles de la déglutition et une voix faible sont notés. Le patient est reconnaissable à sa tête tombante.
Juvénile
La pathologie de Kugelberg-Welander est souvent diagnostiquée après 2 ans. Il est considéré relativement forme légère SMA, de nombreux patients vivent jusqu'à 30 à 40 ans. L'homme est debout, mais il lui est difficile de le faire car muscles faibles. Une atrophie musculaire progressive se produit.
Un enfant jusqu'à 10-12 ans se développe normalement, puis commence à trébucher, à tomber, à perdre la capacité de faire du sport, de courir, de quitter la maison ou simplement de se déplacer sans fauteuil roulant. Le patient souffre de scoliose sévère périodique qui se développe et la forme de la poitrine change.
Ces patients souffrent souvent de fractures et ont une amplitude de mouvement articulaire limitée.
Pathologies tardives
Le quatrième type comprend l'amyotrophie bulbospinale Kennedy, l'amyotrophie distale Duchenne-Aran et l'amyotrophie péronière Vulpian. Les maladies sont généralement diagnostiquées entre 35 et 40 ans, parfois limites d'âge passer de 16 à 60 ans. Le patient constate une perte progressive de la force musculaire, une atténuation des réflexes tendineux et des contractions musculaires visibles.
Dans l'atrophie de Duchenne-Harran, les mains sont principalement touchées. L'amyotrophie vulpienne se reconnaît à la formation d'omoplates en forme d'ailes.

Causes et mécanisme de développement de la maladie
L'amyotrophie spinale se développe en raison d'un gène SMN muté sur le cinquième chromosome. Si les deux parents sont porteurs, il y a 25 % de chances que l'enfant naisse atteint.
Une mutation du gène SMN entraîne une perturbation de la synthèse protéique, entraînant la destruction des motoneurones de la moelle épinière. Influx nerveux ne passe pas aux muscles, qui s'atrophient en raison de l'inactivité, la personne perd la capacité de bouger.
On pense que le tissu musculaire profond perd en premier ses performances.
Diagnostique
La plupart méthode précise La détermination de l'amyotrophie spinale chez les enfants est une analyse ADN. Elle est réalisée aussi bien chez le nouveau-né que pendant développement intra-utérin. De plus, les études suivantes sont réalisées :
- Analyse biochimique. L'objectif est de déterminer le taux d'enzymes : anine aminotransférase, lactate déshydrogénase, créatine kinase. Leur contenu normal permet d'exclure les suspicions de dystrophie musculaire progressive.
- La méthode vise à enregistrer l’activité bioélectrique. La pathologie est caractérisée par un rythme « palissade ».
- IRM. Prescrit pour détecter les signes d'atrophie musculaire.
- Microscopie de la moelle épinière. Il existe des signes de processus dégénératifs dans les cellules des processus nerveux. Elles rétrécissent et gonflent, tandis que les fibres gliales ont une structure dense.
- Spectrométrie de masse en tandem. L'étude permet de clarifier le niveau d'acides aminés et de protéines du SMN.
- Examen histologique muscles striés. Les résultats montreront des groupes de petites fibres.

Si les jeunes envisageant d'avoir un enfant ont des proches atteints d'une pathologie SMA, il leur est recommandé de se soumettre à un examen génétique.
Traitement
L’objectif principal de la recherche visant à traiter l’amyotrophie musculaire spinale est lié à l’augmentation du niveau de protéine SMN. Actuellement médicaments sont en cours de test et officiels Médecine russe ne les utilise pas.
Le traitement comprend aujourd'hui des médicaments qui améliorent le passage des impulsions. C'est Prozerin, Galantamine. Des médicaments nootropiques (Nootropil) sont prescrits, dont la tâche principale est d'améliorer la fonction cérébrale. Les médicaments sont utilisés pour normaliser le métabolisme, par exemple Actovegin. Prescrit biologiquement additifs actifs, aidant à améliorer le métabolisme. Une thérapie vitaminique est indiquée notamment par la prise de vitamines B, C, E. Les stéroïdes anabolisants accélèrent la synthèse des protéines.
Pour la scoliose et autres pathologies de la colonne vertébrale se développant avec la maladie de Dubowitz et de Kugelberg-Welander, il est indiqué correction orthopédique.

Par des méthodes importantes les traitements sont le massage, la physiothérapie, la stimulation neuromusculaire. Une thérapie par l'exercice est prescrite. Exercice physique Ils aident à maintenir la force, par contre, les faire en société, aller à la piscine aide à socialiser et à communiquer avec d'autres personnes.
Il est conseillé aux patients atteints de SMA de suivre un régime. La nourriture est une source de substances nécessaires aux muscles. Ainsi, les acides aminés essentiels se trouvent dans les céréales, la viande, le poisson, les champignons, les noix, produits laitiers fermentés. Les plats recommandés comprennent l'avoine, le blé et le riz brun.
Les épinards, le brocoli, le hareng, les oignons, le pamplemousse et la pastèque aideront à entretenir et à développer naturellement les muscles. Pour augmenter la testostérone, il est recommandé aux hommes de prendre de l'aneth, des panais, du ginseng et du persil.
Prévision
L'évolution de la maladie et la durée de vie de l'enfant dépendent de son type.
Avec l'atrophie de type 1, le pronostic est extrêmement défavorable. Environ 50 % des bébés ne vivent pas jusqu'à deux ans. Pas plus de 10 % des enfants atteints de la maladie de Werdnig-Hoffman peuvent vivre jusqu'à cinq ans. La cause du décès est le plus souvent une pneumonie, un arrêt respiratoire ou un arrêt cardiaque.
Les patients diagnostiqués avec la maladie de Dubowitz vivent en moyenne jusqu'à 10, parfois 12 ans. Environ 30 % des enfants meurent avant l’âge de quatre ans.
Dans la SMA de type III, la mortalité infantile est moins fréquente. Pour de nombreux patients, les symptômes commencent entre la préadolescence et l’adolescence. Après quelques années, ils arrêtent de marcher. De plus, progressivement, une atrophie musculaire est constatée les organes internes, y compris respiratoire.
On pense que la maladie de type IV n’affecte pas l’espérance de vie, mais qu’elle entraîne un handicap.
La prévention
Il n'existe aucune mesure visant à prévenir et à empêcher le développement de la SMA. Une femme qui attend la naissance d'un enfant peut soupçonner un problème en remarquant la faiblesse des mouvements du fœtus. Une analyse ADN peut confirmer ou dissiper les soupçons. Si nécessaire, une commission médicale est tenue qui peut recommander une interruption de grossesse. Le médecin doit parler de la maladie, de son évolution et de ses conséquences.
Après avoir diagnostiqué la maladie chez un enfant déjà né, il est entouré de soins et d'attention. L'utilisation d'un système de ventilation pulmonaire artificielle, d'aspirateurs d'expectorations et de dispositifs spéciaux pour le mouvement d'un bébé qui peut se déplacer contribue à améliorer la qualité de vie et à aider l'enfant à vivre. Il est recommandé de faire régulièrement des massages et de la physiothérapie. Les enfants, même ceux à mobilité réduite, sont emmenés à la piscine.
L'amyotrophie spinale est une pathologie dangereuse mais incurable. Elle se caractérise par une atrophie musculaire. Se produit dans à différents âges. Le pronostic est dans la plupart des cas défavorable.
Les sources suivantes ont été utilisées pour préparer l’article :
Seliverstov Yu. A., Klyushnikov S. A., Illarioshkin S. N. Amyotrophie spinale : concept, diagnostic différentiel, perspectives de traitement // Journal of Nervous Diseases - 2015
Lepesova M.M., Ushakova T.S., Myrzalieva B.D. Diagnostic différentiel amyotrophie musculaire spinale du premier type // Bulletin d'Almaty institut d'état Améliorations des médecins - 2016
Dans quelle mesure l’article a-t-il été utile ?
SauvegarderComme vous avez trouvé cet article utile...
Suivez-nous sur les réseaux sociaux !
Nous sommes désolés que cet article ne vous ait pas été utile !
Améliorons cet article !
L'amyotrophie spinale se manifeste par petite enfance. Les premiers symptômes peuvent apparaître dès 2 à 4 heures âgé d'un mois. Ce maladie héréditaire, qui se caractérise par la mort progressive des cellules nerveuses du tronc cérébral.
Types de problèmes
Selon le moment de l'apparition, la gravité de l'évolution et la nature des changements atrophiques, on distingue plusieurs types de maladie.
L’amyotrophie spinale peut se développer en raison de :
Premier type : aigu (forme Werdig-Hoffman) ;
Le deuxième type : intermédiaire (infantile, chronique) ;
Troisième type : forme Kugelberg-Welander (chronique, juvénile).
Selon les experts, trois types de la même maladie apparaissent en raison de différentes mutations du même gène. L'amyotrophie spinale est maladie auto-immune, ce qui se produit lorsque deux gènes récessifs sont hérités, un de chaque parent. Le site de mutation est situé sur le chromosome 5. Il est présent chez 40 personnes. Le gène est responsable du codage d’une protéine qui assure l’existence des motoneurones dans la moelle épinière. Si ce processus est perturbé, les neurones meurent.
Maladie de Verdin-Hoffman
Vous pouvez suspecter la présence d'un problème même pendant la grossesse. Si un enfant développe une atrophie musculaire spinale de type 1, des mouvements fœtaux lents et tardifs sont souvent observés pendant la grossesse. Après la naissance, les médecins peuvent diagnostiquer une hypotonie musculaire généralisée.

Les atrophies commencent à apparaître dès les premiers mois de la vie. Des lésions fasciculaires du dos, du tronc et des membres sont souvent notées dans les parties proximales. Des troubles bulbaires sont également observés. Ceux-ci incluent une succion lente, des cris faibles et une déglutition altérée. Les enfants atteints de la maladie de Verdin-Hoffman présentent souvent une diminution des réflexes nauséeux, de la toux, du pharynx et du palais. Ils souffrent également de fibrillation des muscles de la langue.
Mais ce ne sont pas tous les signes permettant de déterminer l'amyotrophie musculaire spinale. Symptômes caractéristiques du type 1 de cette maladie, incluent une faiblesse des muscles intercostaux. Dans le même temps, la poitrine des bébés semble aplatie.
Au cours des premiers mois de leur vie, ces enfants souffrent souvent de infections respiratoires, pneumonie et aspirations fréquentes.
Diagnostic de la maladie de Verdin-Hoffman
La maladie peut être déterminée en examinant l'enfant. Il y a un certain nombre signes cliniques, grâce auquel les experts peuvent déterminer que le bébé souffre d'amyotrophie spinale héréditaire. Les diagnostics comprennent :
Test sanguin biochimique (il montrera une légère augmentation de l'aldolase et de la créatine phosphokinase) ;
Étude électromyographique (le rythme palissadique indiquera des lésions des cornes antérieures de la moelle épinière) ;