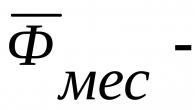Approche scientifique
Objectivement parlant, aucun parent n’est à l’abri de ce risque. Chacun de nous porte en moyenne 10 à 12 gènes défectueux, que nous avons reçus de nos proches et que nous transmettrons peut-être à nos propres enfants. Aujourd'hui, la science connaît environ 5 000 maladies héréditaires qui se développent en raison de problèmes dans l'appareil génétique humain - au niveau des gènes ou des chromosomes.
Ils sont divisés en trois groupes principaux : monogéniques, polygéniques et chromosomiques.
Aujourd’hui, presque toutes les pathologies peuvent s’expliquer d’un point de vue génétique. L'amygdalite chronique est un défaut héréditaire de l'immunité, la lithiase biliaire est un trouble métabolique héréditaire.
Types de maladies
Les maladies monogéniques sont causées par un défaut dans un gène. Aujourd’hui, environ 1 400 de ces maladies sont connues. Bien que leur prévalence soit faible (5 à 10 % du nombre total de maladies héréditaires), elles ne disparaissent pas complètement. Parmi les maladies les plus courantes en Russie figurent la mucoviscidose, la phénylcétonurie, le syndrome surrénogénital et la galactosémie. Pour identifier les pathologies répertoriées, tous les nouveau-nés de notre pays sont soumis à des tests spéciaux (malheureusement, aucun pays au monde ne peut tester les bébés pour la présence de tous les gènes défectueux). Si un écart est détecté, le bébé est transféré à un régime spécial, qui doit être suivi jusqu'à l'âge de 12 ans, et parfois jusqu'à 18 ans. Si des enfants en bonne santé naissent de parents malades, alors tous les descendants de ces derniers seront « sans défaut ».
Les maladies polygéniques (ou multifactorielles) sont associées à une perturbation de l'interaction de plusieurs gènes, ainsi qu'à des facteurs environnementaux. Il s'agit du groupe le plus important - il comprend environ 90 % de toutes les maladies humaines héréditaires, leur prévention et leur traitement ultérieur.
Voies de transmission
Le principal transmetteur de la maladie est une mère ou un père malade. Si les deux souffrent de la maladie, le risque augmente plusieurs fois. En même temps, même si vous et votre conjoint êtes en bonne santé, votre corps contient un certain nombre de gènes défectueux. Ils sont simplement supprimés par les normaux et sont « silencieux ». Si vous et votre mari possédez le même gène « silencieux », vos enfants pourraient développer une maladie héréditaire.
Les maladies « liées au sexe » ont leur propre particularité héréditaire - l'hémophilie, la maladie de Gunther. Ils sont contrôlés par des gènes situés sur le chromosome sexuel. Les parents du patient présentent certains types d'oncologie, des défauts de développement (y compris une fente labiale et une fente palatine). Dans certains cas, les parents ne transmettent pas la maladie elle-même, mais une prédisposition à celle-ci (diabète sucré, maladie coronarienne, alcoolisme). Les enfants reçoivent une combinaison défavorable de gènes qui, dans certaines conditions (stress, blessures graves, mauvais environnement), peuvent conduire au développement de la maladie. De plus, plus la maladie est grave chez maman ou papa, plus le risque est élevé.
Une énorme quantité de temps et d'efforts est consacrée aux maladies héréditaires chromosomiques humaines, à leur prévention et à leur traitement. Elles surviennent en raison de modifications du nombre et de la structure des chromosomes. Par exemple, l'anomalie la plus célèbre - la maladie de Down - est une conséquence du triplement du chromosome 2. De telles mutations ne sont pas si rares ; elles surviennent chez 6 des nouveau-nés. D'autres maladies courantes sont les syndromes de Turner, Edwards et Patau. Tous se caractérisent par de multiples défauts : retard de développement physique, retard mental, défauts des systèmes cardiovasculaire, génito-urinaire, nerveux et autres. Aucun remède aux anomalies chromosomiques n’a encore été trouvé.
l'enfant peut être en bonne santé, mais si la mère est porteuse du gène mutant, la probabilité de donner naissance à un garçon malade est de 5°%. Les filles naissent en bonne santé, mais la moitié d'entre elles deviennent à leur tour porteuses d'un gène défectueux. Un père malade ne transmet pas la maladie à ses fils. Les filles ne peuvent tomber malades que si la mère est porteuse.
D'un tombeau égyptien
Les anciens représentaient le pharaon Akhénaton et la reine Néfertiti avec une apparence plutôt inhabituelle. Il s’avère qu’il ne s’agit pas uniquement de la vision artistique des peintres. En se basant sur la forme anormalement allongée en forme de « tour » du crâne, de petits yeux, de membres anormalement longs (les soi-disant « doigts d'araignée ») et d'un menton indéfini (« visage d'oiseau »), les scientifiques ont identifié le syndrome de Minkowski-Shafar, un des types d'anémie héréditaire (anémie).
De l'histoire russe
Un trouble de la coagulation sanguine (hémophilie) chez le fils du dernier tsar russe Nicolas II, le tsarévitch Alexei, est également de nature héréditaire. Cette maladie se transmet par la lignée maternelle, mais survient exclusivement chez les garçons. Très probablement, la première propriétaire du gène de l’hémophilie était la reine Victoria de Grande-Bretagne, l’arrière-grand-mère d’Alexei.
Difficultés de détection
Les maladies héréditaires n’apparaissent pas toujours dès la naissance. Certains types de retard mental ne deviennent visibles que lorsque l'enfant commence à parler ou commence l'école. Mais la chorée de Gettington (un type de retard mental progressif) ne peut généralement être reconnue qu'après plusieurs années.
De plus, les gènes « silencieux » peuvent aussi devenir des pièges. Leur effet peut se manifester tout au long de la vie - sous l'influence de facteurs externes négatifs (mode de vie malsain, prise d'un certain nombre de médicaments, radiations, pollution de l'environnement). Si votre bébé est à risque, vous pouvez subir un examen de génétique moléculaire, qui permettra d'identifier la probabilité de développer la maladie dans chaque cas spécifique. Ensuite, le spécialiste peut prescrire des mesures préventives. Si les gènes malades s’avèrent dominants, il est impossible d’éviter la maladie. Vous ne pouvez qu'atténuer les symptômes de la maladie. C’est encore mieux d’essayer de les avertir avant l’accouchement.
Groupe à risque
Si vous et votre conjoint présentez l'un des facteurs énumérés, il est préférable de suivre un conseil génétique avant la grossesse.
1. La présence de plusieurs cas de maladies héréditaires sur les deux lignes. Même si vous êtes vous-même en bonne santé, vous pouvez être porteur de gènes défectueux.
2. Âge supérieur à 35 ans. Au fil des années, le nombre de mutations dans l’organisme s’accumule. Le risque de contracter un certain nombre de maladies augmente de façon exponentielle. Ainsi, avec la maladie de Down, pour les mères de 16 ans, c'est 1:1640, pour les 30 ans - 1:720, pour les 40 ans - déjà 1:70.
3. La naissance d'enfants précédents atteints de maladies héréditaires graves.
4. Plusieurs cas de fausses couches. Elles sont souvent causées par de graves anomalies génétiques ou chromosomiques chez le fœtus.
5. Utilisation à long terme par une femme de médicaments (anticonvulsivants, antithyroïdiens, médicaments antitumoraux, corticostéroïdes).
6. Contact avec des substances toxiques et radioactives, ainsi que l'alcoolisme et la toxicomanie. Tout cela peut provoquer des mutations génétiques.
Grâce aux progrès de la médecine, tous les parents ont désormais le choix de perpétuer des antécédents familiaux de maladie grave ou d’y mettre fin.
Méthodes de prévention
Si vous êtes à risque, vous devriez consulter un généticien. Sur la base de votre ascendance détaillée et d’autres données, il décidera si vos inquiétudes sont justifiées. Si votre médecin confirme que vous êtes à risque, vous devez subir des tests génétiques. Il déterminera si vous êtes porteur de défauts dangereux.
Si le risque d'avoir un bébé malade est trop élevé, les experts conseillent de recourir à la fécondation in vitro (FIV) avec diagnostic génétique préimplantatoire (DPI) au lieu de la conception naturelle. Le DPI permet à une cellule prélevée sur un embryon de déterminer s'il est sain ou malade. Seuls les embryons sains sont ensuite sélectionnés et implantés dans l'utérus. Après une FIV, le taux de grossesse est de 40 % (plus d'une procédure peut être nécessaire). Il ne faut pas oublier que l'embryon est testé pour une maladie spécifique (pour laquelle un risque accru est identifié à l'avance). Cela ne signifie pas que l'enfant qui en résulte soit garanti contre d'autres maladies, y compris héréditaires. Le DPI est une méthode complexe et coûteuse, mais elle fonctionne bien entre de bonnes mains.
Pendant la grossesse, vous devez absolument assister à toutes les échographies de routine et donner du sang pour un « triple test » (pour évaluer le degré de risque de développer une pathologie). S'il existe un risque de mutations chromosomiques, vous pouvez subir une biopsie des villosités choriales. Bien qu'il existe un risque d'interruption de grossesse, cette manipulation permet de déterminer la présence d'anomalies chromosomiques. S'ils sont détectés, il est conseillé d'interrompre la grossesse.
Les maladies héréditaires sont des maladies causées par des mutations chromosomiques et génétiques. La science qui étudie les phénomènes d'hérédité et de variabilité dans les populations humaines est la génétique. On croit souvent que les termes « maladie héréditaire » et « maladie congénitale » sont synonymes. Cependant, contrairement aux maladies congénitales qui surviennent à la naissance d’un enfant, les maladies héréditaires sont déjà causées par des facteurs héréditaires et exogènes.
Les problèmes d’hérédité intéressent les hommes depuis des siècles. Par exemple, une maladie telle que l'hémophilie est connue depuis longtemps. À cet égard, les mariages entre parents par le sang étaient interdits. De nombreux scientifiques ont avancé leurs hypothèses sur la survenue de pathologies héréditaires. Leurs hypothèses n'étaient pas toujours fondées sur des observations scientifiques. Ce n’est qu’au XXe siècle, avec le développement de la génétique, que des preuves scientifiques ont été révélées.
Les progrès dans le domaine médical ont conduit à une augmentation relative de la proportion de pathologies d'origine génétique. À ce jour, plus de 3 500 maladies humaines héréditaires ont été identifiées. Environ 5 % des enfants naissent avec des maladies génétiques ou congénitales.
D'un point de vue génétique, toutes les maladies dont le développement est dû à des facteurs héréditaires et environnementaux peuvent être divisées en 3 groupes :
- Maladies héréditaires à mutation phénotypique quasiment indépendantes de l’environnement. Il s'agit généralement de maladies héréditaires génétiques et chromosomiques, telles que l'hémophilie, la maladie de Down, la phénylcétonurie et autres.
- Maladies à prédisposition héréditaire dont la manifestation nécessite l'influence de l'environnement extérieur. Parmi ces maladies figurent le diabète sucré, la goutte, l'athérosclérose, l'ulcère gastroduodénal, le psoriasis, l'hypertension, etc.
- Maladies à l'origine desquelles l'hérédité ne joue aucun rôle. Ceux-ci incluent les blessures, les brûlures et toute maladie infectieuse.
Les maladies causées par des modifications dans la structure des chromosomes sont appelées maladies chromosomiques. Les maladies causées par des modifications de la structure de l’ADN sont appelées maladies génétiques. Le diagnostic clinique des maladies héréditaires repose sur un examen clinique, généalogique et paraclinique.
Il convient de noter que jusqu’à récemment, presque toutes les maladies héréditaires étaient considérées comme incurables. Mais aujourd’hui, tout a changé. En diagnostiquant les maladies à un stade précoce, je peux soulager les souffrances des gens, et parfois même me débarrasser de la maladie. Grâce à la génétique, il existe aujourd'hui de nombreuses méthodes de diagnostic expresses, par exemple les tests biochimiques et les méthodes immunologiques. Un exemple clair est la capacité de la médecine moderne à lutter contre la polio.
Liste des maladies génétiques * Articles détaillés : maladies héréditaires, Maladies métaboliques héréditaires, Enzymopathie. * Dans la plupart des cas, un code est également fourni indiquant le type de mutation et les chromosomes qui y sont associés. aussi... ... Wikipédia
Vous trouverez ci-dessous une liste de rubans symboliques (ruban symbolique ou de notification, du ruban de sensibilisation en anglais), un petit morceau de ruban plié en boucle ; utilisé pour démontrer l'attitude du porteur de la bande face à tout problème ou... ... Wikipédia
Cette page est un glossaire. Voir aussi : Liste des malformations et maladies génétiques Termes génétiques par ordre alphabétique... Wikipédia
Une liste de services d'articles créés pour coordonner les travaux sur le développement du sujet. Cet avertissement n'est pas défini... Wikipédia
Branche de la génétique humaine consacrée à l'étude du rôle des facteurs héréditaires dans la pathologie humaine à tous les niveaux principaux de l'organisation de la vie, de la population à la génétique moléculaire. Section principale de M.g. se compose de génétique clinique,... ... Encyclopédie médicale
Les maladies héréditaires sont des maladies dont l'apparition et le développement sont associés à des défauts de l'appareil de programmation des cellules, hérités des gamètes. Le terme est utilisé en relation avec les maladies polyétiologiques, contrairement à... Wikipédia
Maladies dont l'apparition et le développement sont associés à des défauts de l'appareil de programmation des cellules, hérités des gamètes. Le terme est utilisé en relation avec les maladies polyétiologiques, contrairement au groupe plus restreint Génétique... ... Wikipédia
La maladie héréditaire est une maladie dont l'apparition et le développement sont associés à des défauts de l'appareil de programmation des cellules, hérités des gamètes. Le terme est utilisé en relation avec les maladies polyétiologiques, contrairement à ... ... Wikipedia
Les troubles métaboliques héréditaires comprennent un large groupe de maladies héréditaires affectant les troubles métaboliques. Ces troubles constituent une partie importante du groupe des troubles métaboliques (maladies métaboliques).... ... Wikipedia
Livres
- Maladies infantiles, Belopolsky Yuri Arkadevich. La santé d'un enfant de tout âge est une tâche particulière pour un médecin, car un organisme en croissance nécessite plus d'attention et une plus grande vigilance face aux maladies. Examens médicaux programmés, identification...
- Introduction au diagnostic moléculaire et à la thérapie génique des maladies héréditaires, V. N. Gorbunova, V. S. Baranov. Le livre présente les idées modernes sur la structure du génome humain, les méthodes pour l'étudier, l'étude des gènes dont les mutations conduisent à de graves pathologies héréditaires : considérées...
Maladies héréditaires pédiatres, neurologues, endocrinologues
A-Z A B C D E F G H I J J K L M N O P R S T U V X C CH W SCH E Y Z Toutes les sections Maladies héréditaires Conditions d'urgence Maladies des yeux Maladies des enfants Maladies des hommes Maladies vénériennes Maladies des femmes Maladies de la peau Maladies infectieuses Maladies nerveuses Maladies rhumatismales Maladies urologiques Maladies endocriniennes Maladies immunitaires Maladies allergiques Maladies oncologiques Maladies des veines et des ganglions lymphatiques Maladies des cheveux Maladies dentaires Maladies du sang Maladies du sein Maladies et blessures liées à la SAO Maladies respiratoires Maladies du système digestif Maladies du cœur et des vaisseaux sanguins Maladies du gros intestin Maladies de l'oreille, de la gorge et du nez Problèmes liés aux drogues Troubles mentaux Troubles de la parole Problèmes esthétiques Problèmes esthétiques
Maladies héréditaires– un grand groupe de maladies humaines causées par des modifications pathologiques de l'appareil génétique. Actuellement, plus de 6 000 syndromes à mécanisme de transmission héréditaire sont connus et leur fréquence totale dans la population varie de 0,2 à 4 %. Certaines maladies génétiques ont une prévalence ethnique et géographique spécifique, tandis que d’autres surviennent avec la même fréquence partout dans le monde. L'étude des maladies héréditaires relève avant tout de la responsabilité de la génétique médicale, mais presque tous les médecins spécialistes peuvent rencontrer une telle pathologie : pédiatres, neurologues, endocrinologues, hématologues, thérapeutes, etc.
Les maladies héréditaires doivent être distinguées des pathologies congénitales et familiales. Les maladies congénitales peuvent être causées non seulement par la génétique, mais également par des facteurs exogènes défavorables affectant le développement du fœtus (composés chimiques et médicinaux, rayonnements ionisants, infections intra-utérines, etc.). Dans le même temps, toutes les maladies héréditaires n'apparaissent pas immédiatement après la naissance : par exemple, les signes de la chorée de Huntington apparaissent généralement pour la première fois à l'âge de plus de 40 ans. La différence entre pathologie héréditaire et pathologie familiale est que cette dernière peut être associée non pas à des déterminants génétiques, mais à des déterminants sociaux, quotidiens ou professionnels.
L'apparition de maladies héréditaires est causée par des mutations - des changements soudains dans les propriétés génétiques d'un individu, conduisant à l'apparition de nouvelles caractéristiques inhabituelles. Si des mutations affectent des chromosomes individuels, modifiant leur structure (en raison de la perte, de l'acquisition, de la variation de la position de sections individuelles) ou de leur nombre, ces maladies sont classées comme chromosomiques. Les anomalies chromosomiques les plus courantes sont les pathologies duodénales et allergiques.
Les maladies héréditaires peuvent apparaître immédiatement après la naissance d'un enfant et à différentes étapes de la vie. Certains d'entre eux ont un pronostic défavorable et entraînent une mort précoce, tandis que d'autres n'affectent pas de manière significative la durée ni même la qualité de la vie. Les formes les plus graves de pathologie fœtale héréditaire provoquent un avortement spontané ou s'accompagnent de mortinatalité.
Grâce aux progrès du développement médical, environ un millier de maladies héréditaires peuvent aujourd'hui être détectées avant même la naissance d'un enfant à l'aide de méthodes de diagnostic prénatal. Ces derniers comprennent l'échographie et le dépistage biochimique des trimestres I (10-14 semaines) et II (16-20 semaines), qui sont pratiqués chez toutes les femmes enceintes sans exception. De plus, s'il existe des indications supplémentaires, des procédures invasives peuvent être recommandées : biopsie des villosités choriales, amniocentèse, cordocentèse. Si le fait d'une pathologie héréditaire grave est établi de manière fiable, la femme se voit proposer une interruption artificielle de grossesse pour des raisons médicales.
Tous les nouveau-nés dans les premiers jours de leur vie sont également soumis à un examen pour détecter des maladies métaboliques héréditaires et congénitales (phénylcétonurie, syndrome adrénogénital, hyperplasie surrénalienne congénitale, galactosémie, mucoviscidose). D'autres maladies héréditaires qui n'ont pas été reconnues avant ou immédiatement après la naissance d'un enfant peuvent être détectées à l'aide de méthodes de recherche cytogénétiques, génétiques moléculaires et biochimiques.
Malheureusement, il n’est actuellement pas possible de guérir complètement les maladies héréditaires. Pendant ce temps, avec certaines formes de pathologie génétique, il est possible d'obtenir une prolongation significative de la vie et d'assurer sa qualité acceptable. Dans le traitement des maladies héréditaires, une thérapie pathogénétique et symptomatique est utilisée. L'approche pathogénétique du traitement implique une thérapie de remplacement (par exemple, avec des facteurs de coagulation sanguine dans l'hémophilie), limitant l'utilisation de certains substrats pour la phénylcétonurie, la galactosémie, la maladie du sirop d'érable, comblant le déficit d'une enzyme ou d'une hormone manquante, etc. l'utilisation d'une large gamme de médicaments, de physiothérapie, de cours de rééducation (massage, thérapie par l'exercice). De nombreux patients atteints d'une pathologie génétique dès la petite enfance ont besoin de cours correctionnels et de développement avec un orthophoniste et un orthophoniste.
Les possibilités de traitement chirurgical des maladies héréditaires se réduisent principalement à l'élimination des malformations graves qui interfèrent avec le fonctionnement normal de l'organisme (par exemple, correction des malformations cardiaques congénitales, des fentes labiales et palatines, de l'hypospadias, etc.). La thérapie génique pour les maladies héréditaires est encore de nature plutôt expérimentale et est encore loin d'être largement utilisée en médecine pratique.
La principale direction de la prévention des maladies héréditaires est le conseil génétique médical. Des généticiens expérimentés consulteront un couple marié, prédiront le risque d'avoir une progéniture atteinte d'une pathologie héréditaire et fourniront une assistance professionnelle pour prendre une décision concernant la procréation.
9.1 Concept, classification et caractéristiques de la pathologie héréditaire
La pathologie est tout écart par rapport au déroulement normal des processus biologiques - métabolisme, croissance, développement, reproduction.
La pathologie héréditaire est un écart par rapport à la norme avec un fait établi d'héritage, c'est-à-dire une transmission de génération en génération. Il faut distinguer la pathologie congénitale – présente dès la naissance de l’individu – et la pathologie héréditaire. La pathologie congénitale peut être causée par l'action de facteurs environnementaux - manque de nutriments et d'oxygène pendant le développement intra-utérin, blessures à la naissance, infections, etc. Établir, conformément aux exigences de l'analyse génétique (Chapitre II), le fait de l'hérédité d'un trait anormal est la seule base pour reconnaître le caractère héréditaire de la pathologie.
Il existe deux types de classification des pathologies héréditaires. Le premier (accepté principalement dans la littérature nationale) est le type clinique. Selon ce type de classification, il existe quatre groupes de maladies :
Le groupe I est en fait constitué de maladies héréditaires - maladies chromosomiques et génétiques (syndromes d'Edwards et Patau, phénylcétonurie, mucoviscidose) ;
Groupe II – maladies à prédisposition héréditaire prononcée, dans la pathogenèse desquelles la manifestation de facteurs héréditaires est déterminée par l'action de circonstances extérieures spécifiques (hypertension artérielle, diabète sucré, goutte) ;
Groupe III – maladies déterminées principalement par des facteurs environnementaux, mais dans la pathogenèse desquelles des facteurs héréditaires jouent un rôle (glaucome, athérosclérose, cancer du sein) ;
Groupe IV - maladies avec lesquelles l'hérédité à première vue n'a rien à voir (intoxication alimentaire, fractures, brûlures).
Il convient de noter que les notions fréquemment utilisées de maladies « familiales » et « sporadiques » ne sont pas directement liées à l’hérédité. Les maladies familiales sont observées chez les proches, mais peuvent également être causées par les mêmes causes externes, par exemple la nature de l'alimentation. Des cas sporadiques surviennent chez des individus individuels, mais peuvent également être dus à une rare combinaison d'allèles ou à une mutation de novo.
Le deuxième système de classification - génétique - est généralement accepté dans la littérature étrangère et a récemment trouvé une utilisation de plus en plus fréquente dans la littérature russe. Selon ce système, cinq groupes sont distingués :
Groupe I – maladies génétiques déterminées par des mutations de certains gènes. Il s'agit de traits majoritairement monogéniques avec des modes de transmission autosomique dominant, autosomique récessif, lié au sexe dominant, lié au sexe récessif, holandrique et mitochondrial (Chapitre II) ;
Groupe II – maladies chromosomiques, c'est-à-dire mutations génomiques et chromosomiques (Chapitre V) ;
Groupe III – maladies à prédisposition héréditaire, dans la pathogenèse desquelles jouent un rôle des facteurs environnementaux et héréditaires à transmission de type monogénique ou polygénique (myopie, obésité morbide, ulcère gastrique).
Groupe IV – maladies génétiques des cellules somatiques, souvent associées à des néoplasmes malins (rétinoblastome, tumeur de Wilms, certaines formes de leucémie) ;
Groupe V – maladies d'incompatibilité génétique entre la mère et le fœtus, qui se développent à la suite de la réaction immunitaire de la mère aux antigènes fœtaux (incompatibilité pour le facteur Rh et certains autres systèmes antigène-anticorps érythrocytaires).
Les maladies héréditaires peuvent commencer à se manifester à différents âges. La nature de la manifestation (moment d'apparition des premiers symptômes de la maladie) est spécifique aux différentes formes de pathologie héréditaire. En règle générale, les maladies héréditaires se caractérisent par une évolution chronique (à long terme) progressive (avec une gravité croissante des symptômes).
9.2 Maladies chromosomiques
Ce groupe comprend les maladies causées par des anomalies du nombre ou de la structure des chromosomes. Environ 1 % des nouveau-nés ont un caryotype anormal et chez les mort-nés, l'incidence des aberrations dans le nombre ou la structure des chromosomes est de 20 %. Les caractéristiques communes des maladies chromosomiques sont : un faible poids à la naissance, un retard de développement, une petite taille, une microcéphalie, une micrognathie, des troubles de l'ostéogenèse, une position anormale des yeux. Une description plus détaillée des maladies chromosomiques est donnée dans les sections 5.8 et 5.9.
9.3 Maladies génétiques
Les maladies génétiques sont des états pathologiques provoqués par des mutations génétiques. Le plus souvent, ce concept est appliqué aux maladies monogéniques.
Ce groupe est caractérisé par l'hétérogénéité : les mêmes maladies peuvent être causées par des mutations dans différents gènes. Les principes généraux du développement de la pathologie au niveau génétique peuvent être :
Production d'un produit protéique anormal ;
Manque de protéines normales ;
Quantité insuffisante de protéines normales ;
Excès de produit protéique normal.
Sur la base de la nature des perturbations de l'homéostasie (constance de l'environnement interne de l'organisme), on distingue les groupes de maladies génétiques suivants :
1. Maladies du métabolisme des acides aminés.
Le plus grand groupe de maladies métaboliques héréditaires. Presque tous sont hérités de manière autosomique récessive. La cause des maladies est le déficit de l'une ou l'autre enzyme responsable de la synthèse des acides aminés.
Phénylcétonurie- altération de la conversion de la phénylalanine en tyrosine en raison d'une forte diminution de l'activité de la phénylalanine hydroxylase - une maladie autosomique récessive. Il apparaît vers l'âge de 2-4 mois, les premiers symptômes sont léthargie, crampes, eczéma, odeur de « souris » (odeur de cétones). De graves lésions cérébrales se développent progressivement, entraînant une forte diminution de l'intelligence jusqu'à l'idiotie. Si, dès les premiers jours de la vie, vous excluez complètement (ou limitez considérablement la quantité) la phénylalanine de l'alimentation d'un enfant malade avant la puberté, les symptômes ne se développent pas. La maladie est causée par des mutations du gène HAP, qui code pour la phénylalanine 4-hydroxylase. Gène HAP localisé dans HSA12q24.1. Plusieurs dizaines de mutations de ce gène ont été décrites dans différentes populations. Il existe des systèmes de diagnostic basés sur la PCR qui peuvent détecter le portage hétérozygote. Récemment, de nouvelles approches de traitement de la phénycétonurie ont été développées : thérapie de remplacement par la phénylalanine lyase, une enzyme végétale qui catalyse la dégradation de la phénylalanine en métabolites inoffensifs, et thérapie génique en insérant le gène normal de la phénylalanine hydroxylase dans le génome.
Alcaptonurie– un trouble autosomique récessif du métabolisme de la tyrosine et une accumulation d'acide homogentisique dans les tissus corporels (cartilage articulaire, tendons). La manifestation se produit dans l'enfance. Le premier symptôme est un assombrissement de l’urine. Une lithiase urinaire et une pyélonéphrite se développent souvent. L'accumulation de produits de dégradation de l'acide homogentisique entraîne des lésions articulaires (principalement le genou et la hanche). Il y a un assombrissement et une fragilité accrue du tissu conjonctif. L'assombrissement de la sclère et des oreilles est caractéristique. Mutations dans le gène HGD Les acides homogentisiques oxydases sont à l’origine de cette maladie. Ce gène contient 14 exons et est localisé dans HSA3q21-23. Environ 100 mutations faux-sens, de décalage de cadre et de site d'épissage différentes ont été décrites et sont associées à cette maladie. .
Albinisme oculocutané 1– absence ou déficit important de pigment au niveau de la peau, des cheveux, de l'iris et des membranes pigmentaires de l'œil (Figure IX, 1).
Graphique IX, 1. Un représentant de la race négroïde est un albinos. Basé sur des documents du site http://upload.wikimedia.org/wikipediacommons/99a/Albinisitic_man_portrait
Une maladie avec un type de transmission autosomique récessif. Elle se manifeste par divers degrés de dépigmentation de la peau, des cheveux, de l'iris et des membranes pigmentaires de l'œil, une diminution de l'acuité visuelle, une photophobie, un nystagmus et des coups de soleil fréquents. Diverses mutations faux-sens, frameshift et non-sens dans le gène de la tyrosinase ( TYR, HSA11q24) sont responsables de cette maladie.
2. Troubles du métabolisme des glucides
Galactosémie– absence ou diminution significative de l'activité de l'enzyme galactose-1-phosphate-uridyltransférase et accumulation dans le sang du galactose et de ses dérivés, qui ont un effet toxique sur le système nerveux central, le foie et le cristallin de l'œil. Au cours des premiers jours et semaines de la vie, on observe une jaunisse, une hypertrophie du foie, un nystagmus, une hypotonie musculaire et des vomissements. Au fil du temps, des cataractes et un retard du développement physique et mental se développent. Caractérisé par une intolérance au lait.
La maladie a un mode de transmission autosomique récessif. Plusieurs formes de cette maladie sont causées par différents allèles mutants du gène GALT(galactose-1-phosphate uridyl transférase), localisée dans la région HSA9p13. Les mutations faux-sens réduisent l'activité enzymatique à des degrés divers, ce qui détermine différents degrés de gravité des symptômes de la maladie. Par exemple, la galactosémie de Durthe est presque asymptomatique, seule une tendance aux troubles hépatiques est notée.
Maladie de Gierke (glycogénose de type I, maladie du stockage du glycogène de type I)– incapacité à convertir le glucose-6-phosphate en glucose, ce qui entraîne une perturbation de la synthèse et de la décomposition du glycogène. Le stockage du glycogène se produit, mais pas le processus inverse. Une hypoglycémie se développe. L’accumulation d’un excès de glycogène dans le foie et les reins entraîne une insuffisance hépatique et rénale. Le type de transmission est autosomique récessif. La cause de la maladie est une mutation du gène G6PC, qui code pour l'enzyme glucose-6-phosphatase. Quatorze allèles mutants de ce gène ont été décrits et associés à la maladie de Gierke. Il existe des tests de génétique moléculaire pour identifier le portage hétérozygote et le diagnostic prénatal de cette maladie.
3. Troubles du métabolisme lipidique
Maladie de Niemann-Pick types A et B- diminution de l'activité de l'enzyme sphingomyélinase acide lysosomale, codée par le gène SMPD1(HSA11p15.4-p15.1). Le type de transmission est autosomique récessif. La violation du métabolisme lipidique entraîne l'accumulation de lipides dans le foie, les poumons, la rate et les tissus nerveux. Caractérisé par une dégénérescence des cellules nerveuses, une perturbation du système nerveux, une augmentation des taux de cholestérol et de lipides dans le sang. Le type A est mortel dans la petite enfance. Le type B est plus léger ; les patients survivent généralement jusqu'à l'âge adulte. Différents types sont causés par différentes mutations du gène SMPD1.
Maladie de Gaucher (lipidose glycosylcéramide)- accumulation de glucocérébrosides dans les cellules du système nerveux et réticuloendothélial, provoquée par un déficit de l'enzyme glucocérébrosidase, codée par le gène ACS(HSA1q21). Appartient au groupe des maladies de surcharge lysosomale. Certaines formes de la maladie se manifestent par de graves lésions du foie, de la rate, des tissus nerveux et osseux.
4. Maladies héréditaires du métabolisme des purines et des pyrimidines
Syndrome de Lesch-Nychen – une maladie récessive liée au sexe dans laquelle la teneur en acide urique de tous les fluides corporels augmente fortement. La conséquence en est un retard de développement, un retard mental modéré, des attaques de comportement agressif avec automutilation. Insuffisance de l'activité enzymatique de l'hypoxanthine-guanine phosphoribosyltransférase due à des mutations du gène HPRT1(HSAXq26-q27.2) est à l’origine de cette maladie. Plusieurs mutations dans le même gène ont été décrites, entraînant goutte(altération du métabolisme des purines et dépôt de composés d'acide urique dans les tissus).
5. Troubles métaboliques du tissu conjonctif
Syndrome de Marfan (doigts d'araignée, arachnodactylie)- lésions du tissu conjonctif dues à une mutation du gène FBN1(HSA15q21.1), responsable de la synthèse de la fibrilline. Hérité de manière autosomique dominante. Le polymorphisme clinique de la maladie s'explique par le grand nombre d'allèles mutants, chacun pouvant se manifester à l'état hétérozygote. Les patients se caractérisent par une grande taille, un physique asthénique (membres disproportionnellement longs), une arachnodactylie (doigts longs et fins), une faiblesse de l'appareil ligamentaire, un décollement de rétine, une subluxation du cristallin, un prolapsus de la valvule mitrale (Figure IX, 2).

Figure IX, 2. Le syndrome de Marfan. Basé sur des documents du site http://www.spineinfo.ru/infosources/case/cases_14.html.
Mucopolysaccharidoses- un groupe de maladies du tissu conjonctif associées à une altération du métabolisme des glycosaminoglycanes acides (mucopolysaccharides) provoquée par un déficit de certaines enzymes lysosomales. Ces maladies sont classées comme maladies de surcharge lysosomale. Ils se manifestent par divers défauts des os et du tissu conjonctif. Mucopolysazaridose de type I (syndrome de Hurler) est une maladie autosomique récessive résultant d'un déficit de l'enzyme alpha-L-iduronidase dû à des mutations du gène IDUA (HSA4q16.3). Cela conduit à l'accumulation de complexes protéines-glucides et de graisses dans les cellules du corps. En conséquence, les patients présentent une petite taille, un retard mental important, une hypertrophie du foie et de la rate, des malformations cardiaques, une opacification de la cornée, une déformation osseuse et un grossissement des traits du visage (Figure IX, 3).

Figure IX, 3. Le syndrome de Hurler. Basé sur des documents du site http://medgen.genetics.utah.edu/photographs/pages/hurler_syndrome.htm.
Mucopolysaccharidose de type II(Syndrome de Hunter) est une maladie récessive liée au sexe qui est causée par un défaut de l'enzyme iduronate sulfotase dû à une mutation du gène IDS (HSAXq28). Les substances d'accumulation sont les sulfates de dermatane et d'héparane. Caractérisé par des traits du visage rugueux, une scaphocéphalie, une respiration bruyante, une voix basse et rauque, des infections virales respiratoires aiguës fréquentes (Figure IX, 4 ) . À l'âge de 3-4 ans, des troubles de la coordination des mouvements apparaissent - la démarche devient maladroite, les enfants tombent souvent en marchant. Les patients se caractérisent par une labilité émotionnelle et une agressivité. Sont également observées une surdité progressive, des lésions nodulaires de la peau du dos, de l'arthrose et des lésions cornéennes.
\ 
Graphique IX, 4. Syndrome du chasseur. Basé sur des documents du site http://1nsk.ru/news/russia/23335.html.
Mucopolysaccharidose de type III (syndrome de Sanfilippo, maladie de Sanfilippo) - une maladie causée par l'accumulation d'héparane sulfate. Elle se caractérise par une hétérogénéité génétique - il existe 4 types de cette maladie, provoqués par des mutations dans 4 gènes différents codant pour des enzymes impliquées dans le métabolisme des substances accumulées. Les premiers symptômes de la maladie sous forme de troubles du sommeil apparaissent chez les enfants de plus de 3 ans. L'apathie se développe progressivement, il y a un retard dans le développement psychomoteur, des troubles de la parole et les traits du visage deviennent grossiers. Au fil du temps, les enfants ne reconnaissent plus les autres. Les patients se caractérisent par un retard de croissance, des contractures articulaires, une hypertrichose et une hépatosplénomégalie modérée. Contrairement aux syndromes de Hurler et de Hunter, le retard mental prédomine dans la maladie de Sanfilippo et il n'y a pas de lésions de la cornée et du système cardiovasculaire.

Figure IX, 5. Syndrome de Sanfilippo. Basé sur des documents du site http://runkle-science.wikispaces.com/Sanfilippo-syndrome.
Fibrodysplasie (myosite ossifiante, ossification hétérotopique paraosseuse, maladie de Munheimer)- une maladie du tissu conjonctif associée à son ossification progressive suite à une mutation du gène ACVR1(HSA2q23-q24), qui code pour le récepteur de l'activine A. Le type de transmission est autosomique dominant. La maladie se manifeste par des anomalies congénitales du développement - principalement des gros orteils courbés et des troubles de la colonne cervicale au niveau des vertèbres c2 à c7. La maladie est de nature évolutive, entraînant des altérations significatives de l'état fonctionnel du système musculo-squelettique, un handicap profond des patients et la mort, principalement dans l'enfance et le jeune âge (Figure IX, 6). La maladie est également appelée « maladie du deuxième squelette », car là où les processus anti-inflammatoires normaux devraient se produire dans le corps, la croissance osseuse commence.

Figure IX, 6. Fibrodysplasie. Basé sur des documents du site http://donbass.ua/news/health/2010/02/15.
6. Troubles des protéines circulantes
Hémoglobinopathies- troubles héréditaires de la synthèse de l'hémoglobine. Il existe deux groupes d'hémoglobinopathies. Le premier est caractérisé par une modification de la structure primaire de la protéine globine, qui peut s'accompagner de perturbations dans sa stabilité et sa fonction (par exemple, l'anémie falciforme). Dans les hémoglobinopathies du deuxième groupe, la structure de l'hémoglobine reste normale, seul le taux de synthèse des chaînes de globine est réduit (par exemple, β -thalassémie).
7. Troubles métaboliques des globules rouges
Sphérocytose héréditaire- déficit congénital en lipides membranaires érythrocytaires. La maladie est caractérisée par un mode de transmission autosomique dominant ou autosomique récessif, selon la mutation génétique. SPTA1(HSA1q21), qui code pour la spectrine érythrocytaire α-1. Une anomalie de cette protéine entraîne une augmentation de la concentration d'ions sodium à l'intérieur de l'érythrocyte et la pénétration de l'excès d'eau dans celui-ci en raison d'une augmentation de la pression osmotique. En conséquence, des globules rouges sphériques se forment - des sphérocytes qui, contrairement aux globules rouges normaux biconcaves, n'ont pas la capacité de changer de forme dans des zones étroites du flux sanguin, par exemple lors du passage dans les sinus de la rate. Cela conduit à un ralentissement du mouvement des érythrocytes dans les sinus de la rate et au détachement d'une partie de la membrane érythrocytaire avec formation de microsphérocytes. Les globules rouges détruits sont absorbés par les macrophages de la rate. L'hémolyse des globules rouges entraîne une hyperplasie des cellules pulpaires et une hypertrophie de la rate. L'un des principaux symptômes cliniques est la jaunisse. Les principaux symptômes de la sphérocytose héréditaire sont une hypertrophie de la rate (dépassant généralement de 2 à 3 cm sous l'hypocondre) et une jaunisse. Parfois, il existe des signes de retard de développement, de troubles du squelette facial, d'un crâne en forme de tour, d'un nez en selle, d'un palais haut, d'une violation de la position des dents et d'orbites oculaires étroites.
8. Maladies héréditaires du métabolisme des métaux
Maladie de Konovalov-Wilson (dystrophie hépatocérébrale)– un trouble autosomique récessif du métabolisme du cuivre, entraînant de graves lésions du système nerveux central et des organes internes. La maladie est causée par une synthèse faible ou anormale de céruloplasmine (une protéine de transport du cuivre) en raison d'une activité enzymatique insuffisante de l'ATPase transportant le cuivre. Mutations (environ 200 d'entre elles ont été décrites) dans le gène ATP7B(HSA13q14-q21) entraînent des modifications du β-polypeptide de cette enzyme, qui constitue la base génétique de cette pathologie. Le rôle principal dans la pathogenèse est joué par une violation du métabolisme du cuivre, son accumulation dans les tissus nerveux, rénal, hépatique et cornéen, entraînant des dommages toxiques à ces organes par le cuivre. Une cirrhose à gros nodules ou mixte se forme dans le foie. Dans les reins, les tubules proximaux sont principalement touchés. Dans le cerveau, les noyaux gris centraux, le noyau denté du cervelet et la substance noire sont les plus touchés.
9. Malabsorption dans le tube digestif
Fibrose kystique (mucoviscidose) - une maladie autosomique récessive caractérisée par des lésions des glandes exocrines, un dysfonctionnement grave du système respiratoire et du tractus gastro-intestinal. La cause est des mutations génétiques CFTR(HSA7q31.2), qui code pour un régulateur transmembranaire de la mucoviscidose. La maladie se caractérise par des lésions des glandes exocrines, un dysfonctionnement grave du système respiratoire et du tractus gastro-intestinal.
Intolérance au lactose (hypolactasie) – un état pathologique autosomique récessif de mauvaise absorption du lactose (sucre du lait), dont la base génétique est constituée de mutations dans les régions régulatrices et codantes du gène LCT(HSA2q21), qui code pour la lactase. Cette enzyme est exprimée principalement dans les cellules ciliées de l'intestin et est responsable de la dégradation du lactose en galactose et glucose. Les principaux symptômes d’un déficit en lactase sont les flatulences, les douleurs abdominales, la diarrhée et les vomissements. Chez les enfants, un déficit en lactase peut se manifester par une constipation chronique, de l’agitation et des pleurs après avoir mangé. Dans différentes populations humaines, les fréquences des allèles mutants varient de 1 à 100 %.
10. Troubles hormonaux
Féminisation testiculaire (syndrome de Morris) – une maladie récessive liée au sexe lorsqu'un caryotype masculin (46, XY) manifeste un phénotype féminin. L'expressivité varie. Avec une féminisation incomplète, les gonades se développent selon le type masculin, mais certains caractères sexuels correspondent au sexe féminin avec des degrés de gravité variables - clitoris hypertrophié, fermeture incomplète de la suture scrotale, grandes lèvres scrotales, vagin raccourci (Figure IX, 7) . Avec une féminisation complète, le symptôme principal est l’absence de menstruation et de pilosité sexuelle avec des glandes mammaires bien développées et un phénotype féminin. La cause de la maladie est due à diverses mutations du gène RA(HSAXq11-q12), qui code pour le récepteur des androgènes.

Figure IX, 7. Vue des organes génitaux externes lors d'une féminisation testiculaire incomplète. Basé sur des documents du site http://www.health-ua.org/img/woman/tabl/8_17.jpg.
Syndrome androgénital (pseudohermaphrodisme féminin) – un trouble endocrinien avec un type de transmission autosomique récessif, dans lequel le patient a des organes génitaux externes masculins et une structure hormonale féminine. Les patients ont un clitoris hypertrophié, qui ressemble à un pénis masculin avec une ouverture urogénitale, il n'y a pas d'entrée externe au vagin, les petites lèvres sont absentes et les grandes lèvres ressemblent à un scrotum « coupé ». Dans ce cas, les organes génitaux internes peuvent avoir un aspect normal. La base génétique de la maladie réside dans les mutations génétiques CYP21(HSA6q21.3), qui code pour l'enzyme 21-hydroxylase du groupe du cytochrome P450, impliquée dans la synthèse des hormones aldostérone et cortisol.
9.4 Marqueurs moléculaires dans l'étude de la pathologie héréditaire
Une partie importante des maladies héréditaires et des maladies à prédisposition héréditaire ne sont pas de nature monogénique. Ils peuvent être classés comme traits quantitatifs, c'est-à-dire ceux qui présentent une série continue de variabilité et peuvent être mesurés - par exemple, la taille, le poids, la longueur des membres. Les allèles d'un grand nombre de gènes contribuent à la manifestation de tels traits, c'est pourquoi ils sont appelés polygéniques. Il est possible de retracer leur héritage et d'identifier les gènes dont les allèles sont impliqués dans des processus pathologiques à l'aide de marqueurs génétiques. L'identification de l'hérédité liée (association) de traits phénotypiques avec des marqueurs génétiques permet de retrouver des régions de chromosomes ayant une influence décisive sur les processus étudiés (clonage positionnel) et d'obtenir des systèmes fiables de diagnostic moléculaire (marquage moléculaire). Actuellement, les marqueurs les plus courants en génétique humaine sont les locus microsatellites (Figure IX, 8 ; Section 8.1) et les sites polymorphes mononucléotidiques - SNP (Figure IX, 9), dont les principales caractéristiques sont présentées dans le Tableau IX, 1.
L'analyse de l'expression des gènes (tout ou groupe) sur des biopuces dans des tissus liés à une maladie héréditaire spécifique, dans des conditions normales et pathologiques, permet souvent d'identifier des gènes candidats à la maladie étudiée. La localisation chromosomique des séquences d'ADN affectant un trait quantitatif (QTL) peut être déterminée sur la base du co-héritage avec plusieurs marqueurs rapprochés. S'il est possible de trouver des marqueurs qui limitent le QTL des deux côtés, alors sur la base des données de séquence génomique (sections 7.7 et 8.4), une liste de gènes candidats à la position pour le QTL de la maladie étudiée peut être compilée. En combinant l'analyse de l'expression et les études d'association avec des marqueurs moléculaires, les gènes candidats les plus probables peuvent être identifiés, c'est-à-dire ceux qui apparaîtront sur les deux listes.
Le degré de sensibilité à certains médicaments et l’efficacité de leur utilisation varient considérablement. Pour une même maladie, le médicament adapté à un individu particulier est souvent sélectionné par essais et erreurs. En plus de faire perdre du temps, cette approche entraîne parfois des dommages irréparables à la santé. Actuellement, des systèmes de marqueurs basés sur SNP ont été développés pour un grand nombre de médicaments, permettant a priori (avant l'expérimentation) de prédire la réponse d'un organisme individuel à une substance chimique particulière. Les associations de variantes alléliques individuelles de marqueurs d'ADN avec les caractéristiques des réactions biochimiques constituent la base de la thérapie individuelle (Figure IX, 10).

Figure IX, 8. Dans les locus microsatellites, l'unité de variation est un groupe de nucléotides.

Figure IX, 9. Sur les sites polymorphes mononucléotidiques (SNP), l'unité de variation est un seul nucléotide.
Tableau IX, 1. Comparaison des principales caractéristiques des SNP et des microsatellites.

Figure IX, 10. Le principe de sélection d'une thérapie individuelle basée sur le polymorphisme des répétitions mononucléotidiques - SNP.
Questions de test et devoirs pour le chapitre IX
1. Dans quel groupe de maladies héréditaires peut-on classer la mucoviscidose ?
2. Un hétérozygote peut-il subir une mutation génétique SPTA1 s'agit-il d'une sphérocytose héréditaire ?
3. Quelle maladie héréditaire est causée par l'accumulation d'héparane sulfate ?
4. Pourquoi existe-t-il quatre allèles SNP possibles ?
Lectures supplémentaires pour le chapitre IX
N.P. Bochkov. Génétique clinique // M. : Geotar-Med. 2002. – 457 S.