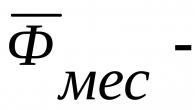24.10.2018
Les maladies fongiques occupent la première place parmi problèmes dermatologiques. Vous pouvez contracter une infection n’importe où. Les maladies de nature fongique peuvent être asymptomatiques ou se manifester de manière plus éloquente - taches sur le corps, démangeaisons, lésions cutanées profondes. Pour soigner les champignons, les médecins prescrivent souvent du Lamisil. Il s'agit de toute une gamme de médicaments dont le principal avantage est leur effet rapide.
Formulaires de décharge
Le nom Lamisil est donné à plusieurs formes posologiques contenant un seul principe actif - la terbinafine. Lamisil est le plus souvent prescrit sous forme de crème. Il est produit en deux emballages - 15 g et 30 g. Le spray Lamisil est populaire. Il s'agit d'une solution liquide à usage externe. Ils sont produits en différents volumes - 15 et 30 ml. Une alternative à la crème est le dermgel. Cette forme galénique est disponible uniquement en doses de 15 g. Pour guérir rapidement le champignon, il existe. médicament spécial– Lamisil Uno. Il s'agit d'une solution cutanée conditionnée dans un tube métallique miniature. En cas de dermatomycose grave, les médecins prescrivent des comprimés de Lamisil. Ils sont vendus par lot de 14 dans une boîte en carton.
Substance active
Ingrédient actif tous les médicaments de la gamme Lamisil-terbinafine. La substance est très différente des autres composés dotés de propriétés antifongiques. C'est caractéristique de lui large éventail action (actif contre les levures, les moisissures et certains champignons dimorphes). La terbinafine tue les agents pathogènes les plus courants du lichen sur le corps et de la dermatomycose sur les pieds - les trichophytons et les microsporums. C'est un composé aux propriétés lipophiles (soluble dans les graisses). Il a taux élevé la biodisponibilité se lie bien aux protéines sanguines et est distribuée dans tous les tissus et organes. La terbinafine s'accumule dans la peau, glandes sébacées, follicules pileux. De ce fait, la substance tue le champignon dans les endroits où les médicaments à usage externe ne peuvent pas toujours pénétrer.
Propriétés pharmacologiques
Les produits Lamisil à usage externe tuent les champignons en surface, à l'intérieur des couches épidermiques (externes) et profondes. Pour augmenter la capacité de pénétration de la terbinafine, tous les médicaments contiennent des conducteurs spéciaux - des alcools organiques.
La solution de Lamisil (sous forme de spray) et le deramgel ont des propriétés à peu près égales. Ils sont bien répartis à la surface de la peau et conviennent au traitement des mycoses cutanées étendues. Les substances ne rendent pas sa surface grasse et sèchent rapidement, ce qui est optimal pour les champignons accompagnés de suintements ou d'érythème fessier (dans les plis de la peau). De plus, ces produits ne tachent pas les vêtements et ne nécessitent pas de frottement, ce qui accélère considérablement leur application.
Creamy Lamisil est prescrit pour les mycoses accompagnées de sécheresse, ainsi que pour les lésions des muqueuses (organes génitaux, oreille externe). Ce médicament possède de légères propriétés hydratantes et élimine l'inconfort associé à l'hyperkératose et à la sécheresse. La crème nécessite un frottement et pénètre moins profondément dans le derme. Les traitements qui l'utilisent sont un peu plus longs qu'avec les solutions.

Lamisil UNO est un médicament antifongique unique. La composition spéciale de la solution, lorsqu'elle est exposée à la peau et à l'air, forme un mince film polymère sur la peau. Il résiste à l'eau, mais se dissout dans le savon et les solvants organiques. Après application, la solution polymérise immédiatement. Cela garantit un contact cutané à long terme avec le médicament. Le composant actif passe du film dans la couche cornée du derme en concentrations élevées. Il tue le champignon et reste actif pendant pendant longtemps. Cette propriété procure un effet préventif pendant 3 mois.
Les comprimés Lamisil sont prescrits pour le traitement des mycoses cutanées et des mycoses des ongles (onychomycose). Les autres formes de Lamisil ne peuvent pas fournir effet thérapeutique avec des dommages aux plaques à ongles. Pour l'onychomycose, il est important que le médicament pénètre dans toutes les couches de l'ongle. Les agents externes Lamisil n'en sont pas capables. Mais les comprimés sont excellents pour lutter contre les dermatomycoses accompagnées d'inflammation.
Les indications
Les indications d'utilisation des préparations externes Lamisil sont les mêmes. Ils sont prescrits pour les dermatomycoses de la peau lisse et parties poilues corps, pieds Les indications les plus courantes comprennent maladies fongiques derme - pied d'athlète inguinal et interdigital, pied d'athlète, teigne et pityriasis versicolor, dermatite séborrhéique. Forme posologique Le médecin sélectionne en fonction de l'intensité des manifestations et des symptômes qui les accompagnent.
Les comprimés sont prescrits pour les infections fongiques des ongles et de la peau sous les cheveux, ainsi que pour toutes les mycoses compliquées ou étendues du corps (dermatomycose des pieds, des jambes, du torse, de l'aine, candidose cutanée).
Les comprimés de Lamisil ne sont pas utilisés pour le pityriasis versicolor (pityriasis versicolor).
Mode d'application
Mode d'emploi du spray Lamisil : A l'aide du distributeur intégré, irriguer généreusement les zones cutanées concernées avec la solution. Il n’est pas nécessaire de frotter le produit avec la main. La solution se répartit bien et sèche rapidement. Pour les maladies cutanées des pieds, du torse et de l'aine, la solution est appliquée une fois par jour (2 fois en cas de symptômes sévères) pendant 7 jours. À pityriasis versicolor le médicament est utilisé pendant 2 semaines.
Lamisil dermagel s'applique sur les zones de dermatomycose avec la paume de la main. Une petite quantité de produit est expulsée du tube sur vos doigts et le gel est réparti uniformément par de légers mouvements de caresses. La fréquence et la durée d'application sont similaires à celles de la pulvérisation.
Mode d'emploi de la pommade Lamisil : pressez une petite quantité de produit sur vos doigts et frottez sur les zones affectées de la peau pendant 3 à 5 minutes. Pour la dermatomycose des pieds, le pityriasis versicolor et la candidose cutanée, le médicament est utilisé 1 à 2 fois par jour (selon les symptômes), pendant 2 semaines. Pour les maladies de la peau lisse du corps, il suffit d'appliquer la crème 7 jours de suite, une fois par jour.

Lamisil Uno s'applique sur les pieds lavés et bien séchés. Vos paumes doivent également être complètement sèches. Environ la moitié du contenu du tube est pressé sur la main et réparti sur le pied, en commençant par l'espace interdigital et les orteils. Arches du pied ( surfaces latérales) sont traités jusqu'à une hauteur de 1,5 cm. Même si les symptômes du champignon ne concernent qu'un seul membre, les deux jambes doivent être lubrifiées. Après répartition du produit, les pieds sont séchés à l'air. Une fois la solution complètement sèche, vous pouvez enfiler des chaussettes. Les pieds ne doivent pas être lavés pendant 24 heures. À l'avenir, prenez soin de vos pieds comme d'habitude, en excluant les frottements avec une pierre ponce, un gant de toilette ou une serviette.
L'avantage du Lamisil externe est un traitement plus court (par rapport à d'autres médicaments).
Les instructions d'utilisation des comprimés doivent être données par un médecin. Souvent, le médicament est prescrit 1 comprimé par jour. Pour les lésions cutanées de la tête, des pieds et du torse, le traitement dure 2 à 4 semaines. Pour l'onychomycose, Lamisil est prescrit pendant 1,5 à 3 mois.
Surdosage

Lors de l’utilisation externe de Lamisil, un surdosage est peu probable. L'ingestion de doses élevées du médicament dans l'organisme est possible en avalant la solution ou le contenu des tubes, car une infime quantité pénètre dans la peau. substance active. Vous pouvez surdoser les comprimés si vous prenez plus de 3 comprimés à la fois.
L'intoxication au Lamisil se manifeste par des nausées, des vomissements et des douleurs abdominales. Le traitement implique un lavage gastrique et la prise de sorbants (charbon actif).
Effets secondaires
Lors de l'utilisation de la crème ou des solutions Lamisil, des réactions de sensibilité locales sont possibles - irritation dans la zone d'application régulière, sécheresse, brûlure, urticaire. Lors de la prise de comprimés par voie orale, des troubles dyspeptiques (troubles des selles, nausées, ballonnements) et un dysfonctionnement des organes hématopoïétiques sont possibles. Lamisil peut perturber le fonctionnement du foie et des reins, pouvant aller jusqu'au développement d'une défaillance de ces organes (avec traitement à long termeà fortes doses).
Contre-indications
Les médicaments de la gamme Lamisil ne sont contre-indiqués que si le patient est individuellement sensible à la terbinafine et aux composants auxiliaires des médicaments. Dans la pratique des enfants, seule la crème est utilisée (à partir de 12 ans). Tous les autres médicaments sont prescrits aux personnes de plus de 18 ans. Les comprimés doivent être recommandés par un médecin. Avant utilisation, il est conseillé d'évaluer l'état les organes internes(en utilisant analyse biochimique sang). En cas de troubles du fonctionnement du cœur, du foie ou des reins, Lamisil est prescrit avec prudence, pour la durée la plus courte possible.
Grossesse et allaitement
Toutes les formes de Lamisil ne sont pas souhaitables pendant l’allaitement. Le médicament pénètre dans la circulation sanguine systémique en petites quantités, mais même celles-ci pénètrent dans le sang. lait maternel. Le médicament peut nuire à la croissance et au développement du nouveau-né. S'il existe un besoin urgent d'un traitement par comprimés, allaitement maternel arrêt.
Femmes enceintes agents antifongiques prescrit que dans des cas extrêmes (par signes vitaux). Les femmes enceintes n'ont pas été incluses dans les essais Lamisil. Les études animales n'ont pas confirmé la capacité de la substance à provoquer des mutations ou des anomalies fœtales. Cependant, il n'existe aucune preuve objective confirmant l'innocuité de Lamisil pour les femmes enceintes.
Analogues
Sur marché pharmaceutique Il existe de nombreux médicaments à base de terbinafine. Ils fonctionnent tous de la même manière que Lamisil. Les produits sont fabriqués par diverses entreprises, ce qui affecte leur prix. Lamisil est l'un des plus chers médicaments antifongiques. Analogues moins chers :
- Vaporisateurs avec terbinafine - Thermikon (240-260 roubles pour 15 ml, 440-460 roubles pour 30 ml), Fungoterbin (420-460 roubles pour 30 ml);
- crèmes– Crème Terbizil (270-310 roubles), Binafin (170-200 roubles), Fungoterbin (320-340 roubles) ;
- pilules– Terbizil (950-1000 roubles), Binafin (620-680 roubles) ; Thermikon (500-550 roubles).

Coût par formulaire de libération
Un grand spray Lamisil (30 ml) coûte 750 à 790 roubles, un petit (15 ml) coûte 590 à 620 roubles. La crème dans un grand tube peut être achetée pour 880 à 920 roubles, dans un petit tube - 550 à 610 roubles. Dermgel Lamisil coûte entre 560 et 600 roubles. 14 comprimés de Lamisil sont vendus en pharmacie pour 2 100 à 2 270 roubles.
Antifongiques systémiques
V.S. Mitrofanov
Les médicaments antifongiques peuvent être classés en fonction des cibles de leur action dans/sur la cellule fongique. Ces classes comprennent : les antibiotiques polyènes, les analogues nucléosidiques (pyrimidines fluorées), les azoles, les pneumocandines-échinocandines, les pradimycines-bénanomycines, les nikkomycines, les allylamines et thiocarbamates, les sordarines et autres (Tableau 1).
Tableau 1.
Mécanismes d'action des médicaments antifongiques.
(Vanden Bossche H, Marichal P. et Odds F. (1994)).
| Cible | Classe chimique | Agent antifongique |
| Synthèse ADN/ARN | pyrimidines | flucytosine |
| Membrane cellulaire
Synthèse de l'égostérol |
Polyènes | Amphotéricine B, nystatine. |
| Squalène époxydase | Allylamines | Naftifine*, terbinafine |
| 14a-déméthylase | Azolés : Imidazoles Triazoles Bistriazoles |
Clotrimazole*, éconazole*, kétoconazole, miconazole.
Fluconazole, itraconazole. Voriconazole, posaconazole |
| D 14 - réductase / D 7 D 8 - isomérase | Morpholines | Amorolfine* |
| Mitose | Griséofulvine | |
| Synthèse du 1,3-b-D-glucane | Échinocandines | Caspofungine |
| Synthèse de chitine | Nikkomycine | Nikkomycine K,Z,T |
| Paroi cellulaire | Pradimycine | BMS-181184 |
| Facteur d'allongement 2 | Soldats | GM-193663, GM-237354 |
* Préparations à usage externe.
Antibiotiques polyènes.
Les antibiotiques polyènes forment des complexes avec l'ergostérol et perturbent la membrane plasmique des cellules fongiques, ce qui entraîne une augmentation de sa perméabilité, une fuite du contenu plasmatique et, par conséquent, la mort de la cellule fongique. Ainsi, les polyènes sont des agents fongicides et possèdent le spectre d'activité antifongique le plus large. L'affinité des polyènes pour l'ergostérol dans les cellules fongiques est nettement plus élevée que pour le cholestérol dans les cellules de mammifères, ce qui rend possible leur utilisation chez l'homme.
Nystatine.
La nystatine a été découverte par Brown et Hazen en 1949 dans des échantillons de sol contenant des actinomycètes. Streptomyces noursei. Elle est utilisée en médecine depuis 1951. Le nom Nystatine signifie l'abréviation N-Y-State (État de New York). Le médicament est mal absorbé par l'intestin après administration per os et il n'est pas administré par voie parentérale. De ce fait, le champ d'application de son application est assez restreint : thérapie locale avec candidose oropharyngée, candidose œsophagienne superficielle, candidose intestinale non invasive.
Amphotéricine-B.
L'amphotéricine B (Amph-B) a été obtenue en 1953. depuis Streptomyces nodosus, isolé par W. Gold et al. à partir d'un échantillon de sol sur le fleuve Orénoque au Venezuela. L'Amph-B est un médicament antifongique à large spectre contre les champignons. Cela a un effet néfaste sur Blastomyces dermatitidis, Coccidioides immitis, Cryptococcus neoformans, Histoplasma capsulatum, Paracoccidioides brasiliensis, Sporotrix spp. Et Candida glabrata. Il est également très actif contre C. albicans et d'autres types Candidose, à l'exclusion C. lusitaniae.
Dans le même temps, Amph-B est variablement actif contre Aspergillus spp. et les zygomycètes ( Mucor spp.), alors que Fusarium, Trichosporon spp. et Pseudoallescheria boydii sont souvent résistants à l’Amph-B. L'administration intraveineuse d'Amph-B reste le traitement principal des mycoses invasives : blastomycose, coccidioïdomycose, paracoccidioïdomycose, histoplasmose, fusarium, méningite cryptococcique (grave et gravité modérée), candidoses, toutes formes d'aspergillose invasive et mucormycose. Le médicament ne pénètre pratiquement pas dans le liquide céphalo-rachidien.
La néphrotoxicité est l’effet secondaire le plus grave de l’Amph-B. Tous les patients recevant Amph-B présentent des troubles fonction rénale plus-moins. L'utilisation d'Amph-B doit être accompagnée d'une surveillance du taux de créatinine et de potassium dans le sérum sanguin. Généralement, lorsque le taux de créatinine dépasse 3,0 à 3,5 mg% (265 à 310 µmol/l), il est recommandé d'interrompre l'administration d'Amph-B pendant plusieurs jours, puis de poursuivre à une dose réduite. Effets indésirables sous Amph-B peut être dose-dépendante (néphrotoxicité, anémie normochrome), idiosyncrasique (rougeur, éruption cutanée, lésion aiguë foie, thrombocytopénie, douleurs générales, convulsions, fibrillation ventriculaire, arrêt cardiaque, fièvre et frissons). Il convient de noter que de la fièvre et des frissons sont observés chez presque tous les patients, tandis que d'autres effets indésirables liés à l'administration d'Amph-B peuvent survenir de manière imprévisible.
Pour réduire le phénomène de fièvre en cours de traitement, Amph-B est parfois prescrit per os acétaminophène (paracétamol) 650 mg toutes les 4 heures ou diphenhydramine (diphenhydramine) 100 mg. Parfois, ces médicaments sont administrés ensemble une demi-heure avant le début de l’administration d’Amph-B. L'administration intraveineuse de prednisolone ou d'hydrocortisone (25 à 50 mg) avant l'administration d'Amph-B réduit également les réactions toxiques. Ces activités sont appelées « prémédication ». Afin de minimiser la toxicité d'Amph-B, une perfusion de 1 litre de solution de chlorure de sodium à 0,9 % a également été utilisée immédiatement avant l'administration d'Amph-B. La méthode la plus efficace pour réduire la toxicité de l’Amph-B est l’utilisation de ses formes liposomales.
Formes d'amphotéricine B associées aux lipides.
Des formes d'Amph-B associées aux lipides ont été développées pour réduire la néphrotoxicité de l'Amph-B traditionnelle. L'Amph-B dans les complexes lipidiques ou les liposomes a une activité antifongique comparable à l'Amph-B traditionnelle, mais diffère par ses propriétés pharmacologiques et toxicologiques. Les complexes lipidiques d'Amph-B (Abelset, AbelcetФ) sont construits comme une membrane double face sous forme de rubans, la dispersion colloïdale d'Amph-B (Amphotec, AmphotecФ, Amphocil, AmphocilD) est constituée de complexes de sulfate de cholestérol avec Amph-B sous forme de disques et de véritables Amph -B liposomaux (Ambisome, AmbisomeФ) - composés sous forme de microsphères (Tableau 2).
Tableau 2.
Caractéristiques des formes d'amphotéricine B associées aux lipides
Nouveaux antibiotiques polyènes.
Il s'agit tout d'abord de la forme liposomale de la nystatine (Nyotran, Nyotran - produite par Aronex), qui, dans l'expérience, a montré une activité élevée contre la candidose invasive et l'aspergillose. La dose efficace variait de 2 à 8 mg/kg. Le principal avantage du niotran est son activité contre toutes les levures résistantes in vitro au fluconazole, à l'itraconazole et aux complexes Amph-B associés aux lipides. Disponible en flacons de 50 mg (en 50 ml) et 100 mg (en 100 ml), le débit de perfusion est de 2 ml/min. Concentration minimale inhibitrice (CMI) in vitro est de 1 µg/ml. Les concentrations sanguines thérapeutiques ont été obtenues après une perfusion unique de nystatine liposomale à la dose de 2 mg/kg.
Le nouveau polyène SPA-S-843 (développé par Societa Prodotti Antibiotici) a montré une activité élevée in vitro contre Candida spp., Cryptococcus spp. Et Saccharomyces spp. et moins toxique que l'Amph-B ordinaire. Également une activité inhibitrice in vitro SPA-S-843 contre. Aspergillus spp..était supérieur à celui de l'Amph-V, et correspondait à l'Amph-V contre R. orizae, P. variotii, Penicillium spp.. Et S. shenkii, mais était inférieur à Amph-B par rapport à Mucor, Microsporium et Trichophyton spp.
Analogues nucléosidiques (pyrimidines fluorées).
La 5-fluorocytosine (flucytosine, ancotyl), un analogue synthétique de la cytosine, a été spécifiquement synthétisée en 1957 pour le traitement de la leucémie, mais en raison du manque de cytotoxicité, elle n'a pas été utilisée à ces fins. L'activité antifongique de la 5-fluorocytosine a été découverte plus tard et prouvée pour la première fois en 1963 dans des modèles expérimentaux de candidose. La 5-fluorocytosine inhibe le métabolisme de la pyrimidine, nécessaire à la synthèse de l'ARN et des protéines dans les cellules fongiques.
Bien que la fluorocytosine soit active in vitro contre Candida spp.. (y compris C. glabrata), Cr. neoformans et Aspergillus spp.., en clinique, il n'était généralement utilisé que pour le traitement de la candidose et de la cryptococcose, ce qui était associé à une faible activité thérapeutique en monothérapie et au développement rapide d'une résistance aux agents pathogènes dans la candidose et la cryptococcose. Malgré le fait que la flucytosine (principalement en association avec l'Amph-B) ait été utilisée pour traiter l'endophtalmie et la méningite à Candida, la méningite cryptococcique et l'aspergillose invasive, en raison de l'avènement de nouveaux médicaments antifongiques, elle n'est désormais pratiquement plus utilisée.
Dérivés azolés.
Initialement, les dérivés azolés comprenaient les imidazoles (clotrimazole, miconazole et kétoconazole), suivis des triazoles de 1ère génération (fluconazole et itraconazole), puis des dérivés du fluconazole de 2ème génération (voriconazole, ravuconazole) et de l'itraconazole (posaconazole).
Les azoles inhibent l'enzyme C14-a des champignons, une déméthylase du système du cytochrome P450, responsable de la conversion du lanostérol en ergostérol. Cela conduit à une diminution de l'ergostérol dans la membrane cellulaire fongique et à sa mort. Activité in vitro pour les azoles varie et ne coïncide pas toujours avec l’activité clinique. Les azoles sont actifs contre C. albicans, Cryptococcus neoformans, Coccidioides immitis, Histoplasma capsulatum, Blastomyces dermatitidis, Paraccoccidioides brasiliensis; généralement résistant aux azoles Candida glabrata, Aspergillus spp., Fusarium spp. et les zygomycètes (Tableau 3).
Tableau 3.
Spectre d'activité des azoles antifongiques
| Agent pathogène | Kétoconazole | Itraconazole | Fluconazole |
| Candida albicans | ++ | +++ | ++++ |
| C. tropicale | ++ | ++ | ++ |
| C. krusei | + | ++ | + |
| C. glabrata | + | ++ | + |
| C. parapsilose | ++ | +++ | ++++ |
| Cryptococcus néoformans | + | ++ | +++ |
| Aspergillus spp. | 0 | +++ | 0 |
| Fusarium spp. | 0 | b | b |
| Pseudallescheria boidii | + | +++ | ++ |
| Classe Zygomycètes | 0 | 0 | 0 |
| Exc. phaeohyphomycose | + | +++ | + |
| Histoplasma capsulatum | ++ | ++++ | +++ |
| Blastomyces dermatitidis | ++ | +++ | + |
| Coccidioides immitis | ++ | +++ | +++ |
| Sporothrix schenckii | + | ++++ | ++ |
| Paracoccidioides brasiliensis | +++ | ++++ | ++ |
| Pénicillium marneffei | + | ++++ | + |
(En utilisant les données de Graybill J.R., 1989)
Les plus anciens (premiers) azoles.
Le clotrimazole et le miconazaol, découverts en 1969, sont mal absorbés lorsqu'ils sont pris par os, Cependant, le clotrimazole ne peut pas être administré par voie parentérale et est utilisé presque exclusivement pour le traitement local des candidoses buccales et vaginales. À une certaine époque, des préparations de miconazole à usage intraveineux (Daktarin) étaient produites, mais leur effet n'a pas été jugé tout à fait optimal et le miconazole est principalement utilisé pour le traitement des mycoses superficielles.
Azolés systémiques actuellement utilisés, dont le voriconazole, qui seront largement disponibles pratique clinique dans un futur proche, sont présentés dans le tableau 4
Tableau 4
Pharmacocinétique comparative des azoles
| Possibilités | Kétoconazole | Itraconazole | Fluconazole | Voriconazole |
| Max. conc. après avoir pris 200 mg (mcg/ml) | 3-5 | 1,0 | 10 | 1-2,5 |
| Autorisation | foie | foie | reins | foie |
| Linéarité | Oui | Non | Oui | Non* |
| Demi-vie | 1-4 | 21-37 | 27-37 | 6-24 |
| Introduction | Per os | Per os | Par os/vv | Par os/vv |
| Effets sur l'absorption en cas de prise per os :
Acidité De la nourriture grasse |
+++ | ++ | 0 | Accepter avec l'estomac vide |
| Pénétration (% sérum) | ||||
| Urine | 2-4 | <1 | 80 | 5 |
| Alcool | <10 | <1 | 50-90 | 50 |
*Note. La pharmacocinétique du voriconazole est non linéaire après l'administration per os, est linéaire jusqu'à 4 mg/kg lorsqu'il est administré par voie intraveineuse, mais après 4 mg/kg, il devient non linéaire (augmente de manière disproportionnée).
Kétoconazole (Nizoral) )
Le kétoconazole, découvert en 1978, présente une bonne absorption orale, un large spectre d'activité et une faible toxicité, mais peut être hépatotoxique et provoquer certains troubles dyshormonaux, comme une diminution des taux de testostérone et de la synthèse d'ACTH. Il n’existe aucune forme posologique de kétoconazole pour l’administration intraveineuse. Le kétoconazole oral est efficace chez les patients atteints de candidose, de coccidioïdomycose, de blastomycose, d'histoplasmose, de paracoccidioïdomycose et de dermatophytose. Le kétoconazole se lie aux protéines, pénètre mal à travers la barrière hémato-encéphalique et n'est pas utilisé pour traiter les lésions du SNC. Le kétoconazole provoque une hépatite dans environ 5 % des cas.La dose de kétoconazole - 200 à 400 mg par jour pendant 5 à 7 jours n'affecte pas la pharmacocinétique de l'aminophylline. Cependant, dans d'autres études, une augmentation de la teneur en théophylline de 22 % a été notée. Actuellement, il est remplacé dans la pratique clinique par des azoles de deuxième génération. Le kétoconazole est pris avec de la nourriture, ce qui garantit son absorption maximale. Le médicament peut être lavé avec du Coca-Cola ou de l'eau de Seltz et, dans certains cas, il est dissous dans de l'acide chlorhydrique, du suc gastrique ou avec de l'acidine-pepsine et bu avec une paille afin de ne pas endommager les dents avec de l'acide.
Fluconazole (Diflucan) ) .
Le fluconazole a été découvert en 1981. Il s'agit d'un bistriazole métaboliquement stable, hydrosoluble et faiblement lipophile qui se lie mal aux protéines plasmatiques. Le médicament est actif par voie orale et intraveineuse, et les deux voies ont une pharmacocinétique identique. Par exemple, l'administration une fois par jour de fluconazole entraîne des concentrations élevées et un équilibrage rapide du médicament dans les tissus corporels avec une bonne disponibilité tissulaire, y compris une pénétration dans le liquide céphalo-rachidien, par ex. 100 mg par jour, la concentration sérique est de 4,5 à 8 mcg/mlc 89% pénétration dans le liquide céphalo-rachidien. Le fluconazole est bien toléré, présente un très faible niveau d'effets secondaires et une large gamme d'activités antifongiques, à l'exclusion des champignons du genreAspergillus spp.. Il convient de noter qu'il existe des différences significatives dans l'activité du médicament contre les champignons dans les modèlesin vivo Et in vitro, qu'il faut garder à l'esprit lors du choix d'un traitement antifongique en tenant compte de la sensibilité. Données d'activité comparativesin vitro Et in vivo contre Candida albicanspour les azoles et l'Amph-B sont présentés dans le tableau 5.
Tableau 5.
Données comparatives sur l'activité des médicaments antifongiques in vitro(MIC) et in vivo(concentration minimale efficace mg/kgj 4) .
L'absorption du fluconazole ne dépend pas du pH gastrique ni de la prise alimentaire. Il est très soluble dans l’eau, c’est pourquoi il est disponible pour une administration intraveineuse. Le fluconazole est unique parmi les agents antifongiques connus dans la mesure où il est excrété par les reins principalement sous forme inchangée (69 à 90 %) et seulement environ 4 % dans l'urine sous forme de métabolite. Les métabolites du fluconazole actifs contre les champignons sont inconnus. Le médicament s'accumule dans les tissus pendant 2 semaines maximum. Le fluconazole est librement sécrété par la salive et les liquides alimentaires, ce qui est prouvé par l'éradication Candida spp. des intestins avec administration intraveineuse. Résistant au fluconazole C. krusei Et C. glabrata.
Il a été démontré que le fluconazole interagit avec le CYP2C9 et le CYP3A4 du système du cytochrome P450, mais il s'agit d'un inhibiteur significativement plus faible du CYP3A4 que les autres azoles, comme l'ont montré des expériences avec la cyclosporine. Parallèlement, il réduit encore la clairance de la cyclosporine et de la warfarine, ce qui doit être pris en compte lors de leur utilisation conjointe. Aucun inhibiteur cliniquement significatif du métabolisme du fluconazole n'a été identifié chez l'homme, mais son niveau peut être affecté par des médicaments excrétés par les reins et affectant la clairance rénale. D'autre part, la cimétidine, étant un inhibiteur du cytochrome P450, réduit la concentration plasmatique du fluconazole de 20 %, ce qui est probablement dû à une diminution de l'absorption. Des rechutes d'infections mycosiques et une diminution de l'ASC (aire sous la courbe) du fluconazole lorsqu'il est pris en association avec la rifampicine et le fluconazole ont été décrites.
Actuellement, le fluconazole est l'un des médicaments les plus efficaces pour le traitement des candidoses oropharyngées, œsophagiennes et vaginales, en particulier chez les patients atteints d'une infection par le VIH ou d'un cancer. Il est également efficace pour la péritonite, la candidémie ou la candidose disséminée (y compris les processus chez les patients atteints de neutropénie), la candidose hépatosplénique ; et est le principal médicament contre la candidurie et d'autres lésions du système urinaire. L'administration orale à long terme de fluconazole après un traitement par Amph-B prévient les rechutes d'endocardite à candidose. Le fluconazole a été utilisé avec succès pour traiter la cryptococcose pulmonaire et disséminée, en particulier chez les patients infectés par le VIH. Le fluconazole 200 mg trois fois par semaine chez les patients infectés par le VIH ayant un nombre de CD4 inférieur à 100 s'est avéré efficace pour la prévention primaire de l'infection cryptococcique. Le fluconazole est bien toléré, même à des doses très élevées telles que 2 000 mg par jour.
Itraconazole (Orungal)
L'itraconazole, découvert en 1986, est un triazole doté d'un large spectre d'activité antifongique, incluant les champignons du genre Aspergille. Il est peu soluble dans l’eau et n’est actuellement disponible que pour une administration orale. Le médicament peut être administré une fois par jour. Cependant, des doses élevées (plus de 400 mg/jour), utilisées dans les processus mycotiques sévères et la thérapie par impulsions, sont prescrites en deux doses. En raison du caractère lipophile de l'itraconazole, sa concentration dans la peau peut être de 10 et dans le foie, de 10 à 20 fois supérieure à celle du plasma sanguin. La biodisponibilité de l'itraconazole peut varier considérablement et est plus élevée lorsque le médicament est administré avec de la nourriture. La prise de jus de pamplemousse, qui est un inhibiteur alimentaire du cytochrome C450, n'affecte pas la pharmacocinétique de l'itraconazole. L'itraconazole est largement métabolisé chez l'homme : aucun médicament inchangé n'a été trouvé dans l'urine et moins de 20 % ont été trouvés dans les selles. Généralement, l'itraconazole est métabolisé en métabolite actif p-hydroxyitraconazole, qui est un métabolite important en raison de son activité antifongique, bien que moindre. que celui de l'itraconazole, ainsi qu'en raison de la tendance à s'accumuler dans le sérum sanguin à des concentrations élevées.
L'ASC de l'itraconazole après une dose de 200 mg était environ dix fois supérieure à celle après une dose de 50 mg. Le métabolisme principal de l’itraconazole s’effectue via l’isoenzyme CYP3A4. Cependant, de nombreux médicaments affectés par l'itraconazole sont des substrats de la glycoprotéine P, qui assure le transport du médicament dans l'intestin grêle, puisque l'itraconazole est un inhibiteur de l'activité de la glycoprotéine P. La cimétidine réduit la demi-vie de l'itraconazole de 40 %. L'itraconazole ne doit pas être prescrit simultanément avec des antiacides, des médicaments anticholinergiques, des antagonistes des récepteurs de l'histamine H2, de l'oméprazole, car une augmentation du pH gastrique entraîne une diminution de l'absorption de l'itraconazole. Ce qui précède s'applique à l'itraconazole, produit en gélules. L'utilisation de l'itraconazole en mélange avec la b-hydroxycyclodextrine a permis de créer des formes pour administration intraveineuse et en même temps d'obtenir une absorption supérieure à 60 % lors de la prise. per os Actuellement, l'itraconazole est produit sous forme de solution buvable (10 mg par ml, 200 mg par flacon). La dose habituelle est de 10 ml (100 mg) à jeun. Les formulations intraveineuses d'itraconazole font l'objet d'essais cliniques.
L'absorption de l'itraconazole est réduite chez les patients atteints de leucémie aiguë et d'infection par le VIH. Bien qu'il n'y ait pas de corrélation claire entre la réponse clinique et les concentrations sériques d'itraconazole, la surveillance des concentrations sériques chez les patients gravement malades est nécessaire pour contrôler l'absorption orale. La faisabilité de prescrire des « doses dites de saturation » (300 mg deux fois par jour - 3 jours) est possible pour certains groupes de patients. Les concentrations d'itraconazole dans le liquide céphalo-rachidien, les yeux et la salive sont négligeables.
La prise avec l'astémizole, le cisapride, la terbénafine est dangereuse en raison de la possibilité d'arythmies cardiaques. S'il est nécessaire de prescrire des antihistaminiques, il est conseillé d'utiliser les métabolites actifs de la terfénadine (texofénadine) et de l'hydrocysine (cétirizine).
Métabolisme et interactions médicamenteuses des azoles antifongiques.
Tous les azoles antifongiques sont métabolisés à l'aide du système du cytochrome P450. Le système du cytochrome P450 fait référence à un groupe d'isoenzymes contenant de l'hème (CYP) situées sur la membrane du réticulum endoplasmique lisse, principalement dans le foie et l'intestin grêle.
Le système isoenzymatique du cytochrome P450 joue un rôle important dans le métabolisme de nombreuses substances endogènes (stéroïdes, hormones, prostaglandines, lipides et acides gras) et dans la détoxification des composants endogènes (notamment après administration orale). Tous les médicaments peuvent être divisés en trois groupes en fonction du système du cytochrome P450 : substrats, inducteurs et inhibiteurs de ce système.
Les substrats sont des médicaments métabolisés par l'action catalytique des enzymes du système du cytochrome P540. La plupart des médicaments sont métabolisés principalement par une seule enzyme P450. Le kétoconazole et l'itraconazole sont des substrats du système du cytochrome P450.
Que sont les inhibiteurs du P450 ? Ce sont des médicaments qui suppriment le métabolisme des substrats P450 ; le processus est compétitif et réversible : dès que l'inhibiteur est retiré, le métabolisme revient à la normale. Les médicaments peuvent ne pas être des substrats et peuvent être des inhibiteurs du P450. Par exemple, le fluconazole est un faible inhibiteur du P450, mais ce n’est pas un substrat du P450 et il est principalement excrété par les reins. Le kétoconazole et l'itraconazole, au contraire, sont des inhibiteurs prononcés du système du cytochrome P450.
Que sont les inducteurs P450 ? Les médicaments inducteurs augmentent le nombre d'isoenzymes P450 in vivo. Ce processus est associé à l'activation de la synthèse enzymatique. Contrairement à l’action des inhibiteurs, l’induction dure plusieurs jours même après l’arrêt du médicament inducteur. La rifampicine et le phénobarbital sont les deux inducteurs les plus puissants de la synthèse de l'enzyme P450. Parmi les médicaments antifongiques, la griséofulvine est un inducteur du P450.
La plupart des médicaments sont éliminés de l’organisme par le foie et les reins. Seul un petit nombre d’entre eux sont excrétés d’une autre manière. De très grosses macromolécules, telles que l'héparine et l'Amph-B, sont absorbées par les cellules phagocytaires telles que les cellules de Kupffer du foie. Cette voie est appelée clairance réticuloendothéliale.
Les trois azoles (kétoconazole, fluconazole et itraconazole) utilisés dans le traitement antifongique peuvent bloquer le métabolisme des médicaments qui utilisent l'isoenzyme CYP3A4 comme substrat du métabolisme (c'est-à-dire l'astémizole, la terfénadine, la loratadine, le cisapride, la cyclosporine, l'érythromycine, la clarithromycine, l'oméprazole) . Par exemple, 99 % de la terfénadine entrant dans l’organisme est métabolisée par l’isoenzyme CYP3A4. Cette isoenzyme présente une variabilité d'expression significative et est responsable de 10 à 60 % de l'activité totale du cytochrome P450 dans le foie. Le kétoconazole et l'itraconazole peuvent provoquer un allongement de l'intervalle QT sur l'ECG lorsqu'ils sont utilisés avec l'astémizole et la terfénadine. La loratadine est également métabolisée par le système hépatique du cytochrome P450, le CYP3A4, mais en présence d'inhibiteurs du CYP3A4, elle peut être métabolisée via une voie alternative via le CYP2D6. Le kétoconazole (200 mg 2 fois par jour pendant 5 jours) a inhibé le métabolisme de la loratadine chez des personnes apparemment en bonne santé. Des rapports font également état d'un lien possible entre l'utilisation de loratadine et la survenue d'arythmies cardiaques. La combinaison la plus sûre lors de l'utilisation d'azoles antifongiques avec des antihistaminiques est l'utilisation de texofénadine (Telfast) ou de cétirizine (Zyrtec). Tous les antifongiques azolés peuvent potentialiser la cardiotoxicité lorsqu'ils sont utilisés avec le cisapride (bien que le fluconazole n'ait pas contribué à la cardiotoxicité lorsqu'il est administré avec l'astémizole et la terfénadine). Les azoles antifongiques peuvent renforcer l'effet de la warfarine et augmenter considérablement les taux de cyclosporine. L'association de la cyclosporine avec ces trois médicaments nécessite donc une surveillance de ses concentrations sériques.
Étant donné que les triazoles inhibent le CYP3A4, l'une des enzymes responsables du métabolisme de la théophylline, leur co-administration peut entraîner une augmentation des taux de théophylline. Une toxicité importante de la théophylline peut survenir pendant le traitement par le fluconazole. Les taux de théophylline peuvent augmenter, diminuer ou ne présenter aucun changement significatif lors de la prise de kétoconazole, probablement parce que la théophylline est métabolisée par plusieurs isoenzymes P450. Les taux de théophylline doivent donc être surveillés pendant le traitement par le kétoconazole.
Dans les situations ci-dessus, la terbinafine est une alternative sûre et peut être utilisée pour remplacer le kétoconazole, le fluconazole ou l'itraconazole. Si le remplacement des médicaments ne peut pas être utilisé, il est nécessaire de surveiller leur toxicité. Les principales interactions médicamenteuses des azoles sont présentées dans le tableau 6.
Tableau 6
Interactions médicamenteuses des azoles antifongiques
(Lasar J.D. et coll. (1990); Como J.A. et coll. (1994).
| Une drogue | Kétoconazole | Itraconazole | Fluconazole |
| Augmenter la clairance des azoles | |||
| Rifampicine | ++++ | ++++ | ++ |
| Rifabutine | +++ | + | |
| Phénytoïne | +++ | +++ | 0 |
| Isoniazide | +++ | 0 | 0 |
| Les niveaux de médicaments augmentent lorsqu’ils sont pris avec des azoles. | |||
| Phénytoïne | ++ | ++ | + |
| Carbamazépine | ++ | ++ | + |
| Warfarine | ++ | ++ | + |
| Cyclosporine | +++ | +++ | + |
| Terfénadine | +++ | ++ | + |
| Astémizole | ++ | ++ | ? |
| Sulfonylurées | + | + | + |
| Digoxine | + | + | + |
| Réduire les niveaux d’azole | |||
| Clarithromycine | + | ||
Note:
Effet très prononcé sur la concentration du médicament (l'association est inefficace)
Influence prononcée (forte probabilité d'effets secondaires)
Impact important (effets secondaires possibles)
Faible impact (à considérer)
0 - aucune interaction
Aucune information sur les interactions médicamenteuses
Développements prometteurs des azoles.
Les azoles antifongiques sont en plein développement, parmi lesquels seul le voriconazole est actuellement introduit dans la pratique clinique.
Voriconazole.
Le voriconazole, créé en 1995, est un dérivé du fluconazole. Il est dix fois plus actif que le fluconazole pour agir contre Aspergille spp ., Cryptocoque spp . Et Candidose spp., y compris C. krusei Et S. glabrata résistant au fluconazole. De plus, le voriconazole a montré une activité non seulement fongistatique, mais également fongicide contre Aspergille spp. à des concentrations environ deux fois supérieures à la CMI. Activité in vitroétabli pour les agents pathogènes endémiques ( Blastomyces dermatitidis, Coccidioides immitis, Paracoccidioides brasiliensis Et Histoplasma capsulatum), ainsi que des agents pathogènes potentiels, notamment Fusarium spp., Acremonium kilensii, Scedosporium infatum, Trichosporon spp. Et Pseudallescheria boydii résistant au fluconazole, à l'itraconazole et à l'Amph-B. Le voriconazole est produit sous des formes posologiques à usage oral et intraveineux, pénètre bien dans les tissus corporels, y compris le cerveau et le liquide céphalo-rachidien, et présente un faible niveau d'effets secondaires. La biodisponibilité du voriconazole est supérieure à 80 %, mais il convient de garder à l'esprit que la prise du médicament dans l'heure qui suit un repas la réduit. Lorsqu'elle pénètre dans l'organisme, 60 % de la substance active se lie aux protéines du sérum sanguin. Le métabolisme s'effectue via le système du cytochrome P450 : les isoenzymes CYP2C9, CYP3A4 et CYP 2C19. Le voriconazole peut inhiber l'activité du CYP2C9, du CYP2C19 et, dans une moindre mesure, du CYP3A4.
Posaconazole
Le posaconazole (SCH -56592) est un triazole de deuxième génération et un analogue structurel de l'itraconazole. Le médicament a une faible solubilité dans l'eau (moins de 2 mg/ml) ; il est produit uniquement pour usage oral (en comprimés à 100 mg et en suspension buvable). Niveau d'inhibition de la C14a - déméthylase dans A. flavus Et A. fumigatus pour le posaconazole est 10 fois plus élevé que pour l’itraconazole. La demi-vie variait entre 15 et 25 heures et dépendait de la dose. Le médicament pénètre mal dans le liquide céphalo-rachidien, cependant, certains effets positifs ont été notés sur les lésions du système nerveux central. Les modèles expérimentaux ont montré une grande efficacité contre Coccidioides immitis. Des études animales ont montré qu'atteindre des concentrations plasmatiques de posaconazole de 1 à 2 mcg/ml était efficace pour éradiquer la plupart des infections fongiques systémiques mortelles. Les effets secondaires comprennent des étourdissements, des maux de tête et de la somnolence.
Ravuconazole.
Le raviconazole (BMS-207147), un dérivé du fluconazole, a montré une activité élevée in vitro et une efficacité élevée dans les modèles expérimentaux d'aspergillose invasive, comparables à l'Amph-B, ainsi qu'une activité plus élevée que l'itraconazole et le fluconazole contre Candidose spp . (y compris C. krusei), Coccidioides, Histoplasma, Fusarium Et Blastomyces par rapport à l'itraconazole et au fluconazole, en maintenant des concentrations fongicides proches de la CMI. Il était également supérieur au fluconazole dans les modèles in vivo avec cryptococcose et candidose du tractus gastro-intestinal. La demi-vie était très longue, allant de 5 à 8 jours, avec une bonne biodisponibilité et tolérabilité. C'est la longue demi-vie qui nécessite une étude en termes d'effets et d'interactions médicamenteuses, puisque, selon d'autres données, dans l'aspergillose invasive expérimentale chez le lapin, la demi-vie était de 13 heures et aucune accumulation du médicament n'a été notée 6 jours après l'arrêt du traitement.
Échinocandines et pneumocandines
Les échinocandines sont des agents fongicides lipoprotéiques cycliques qui interfèrent avec la synthèse de la paroi cellulaire en raison de l'inhibition non compétitive de la synthèse du 1,3-b-D-glucane, une enzyme absente chez les mammifères. Cette inhibition est très spécifique et même une brève exposition au médicament entraîne la mort de la cellule fongique. L'inconvénient des échinocandines est leur faible activité contre les cryptocoques. Les pneumocandines sont des analogues des échinocandines (une des classes de lipoprotéines d'échinocandine). Le nom de « pneumocandines » est dû au fait qu'elles ont une activité contre Pneumocystis carinii, et aussi contre Candidose Et Aspergillus spp.. Comme d’autres analogues de l’échinocandine, les pneumocandines ont peu d’activité contre les cryptocoques.
Le premier médicament de cette classe dont l'utilisation est approuvée est la caspofungine (Cancidas, CancidasF, MK-0991) de Merck, produite sous forme posologique pour administration intraveineuse (le flacon contient 50 mg du médicament, dilué dans une solution de chlorure de sodium à 0,9 %). . Le médicament est principalement destiné au traitement antifongique des patients atteints de formes invasives d'aspergillose, résistantes au traitement standard ou intolérantes à d'autres médicaments antifongiques. Doses recommandées : le premier jour, 70 mg une fois, puis 50 mg par voie intraveineuse une fois par jour. Recherche in vitro ont montré que la caspofungine n'est ni un inhibiteur ni un substrat d'aucune enzyme du système du cytochrome P450. Des études sur des volontaires sains ont montré que la caspofungine n'interagit pas avec d'autres médicaments antifongiques (itraconazole ou Amph-B). Lorsque la caspofungine est prescrite avec des inducteurs de clairance du médicament tels que la rifampicine, la dexaméthasone, la carbamazépine, la dose de caspofungine peut être augmentée jusqu'à 70 mg en l'absence de réponse clinique adéquate. Il n'existe aucune donnée sur la possibilité d'utiliser la caspofungine en parallèle avec la cyclosporine, cette association n'est donc pas encore recommandée. Les effets secondaires comprenaient de la fièvre, une phlébite, une thrombophlébite au site de perfusion, des maux de tête, des nausées, des éruptions cutanées, un érythème cutané, une légère élévation des enzymes hépatiques et des cas d'anaphylaxie (informations sur le fabricant - www.merck.com).
D'autres médicaments de cette classe, l'anidulafungine (V-échinocandine, fabriquée par Versicor) et la micafungine (FK-463, fabriquée par Fujisawa), en sont à la dernière étape des essais cliniques.
Pradimycines et bénanomycines.
Les pradimycines et les bénanomycines sont des composants fongicides qui se lient selon un mécanisme dépendant du calcium aux mannoprotéines de la paroi cellulaire, ce qui provoque une lyse osmotique et une fuite des composants intracellulaires, conduisant à la mort de la cellule fongique. Aucun effet dépendant du calcium sur les cellules de mammifères n’a été détecté dans ces classes d’agents antifongiques. Les pradimycines-bénanomycines sont fongicides contre de nombreux champignons, y compris ceux résistants à d'autres agents antifongiques. Le BMS-181184 s'est révélé efficace, bien que moins efficace que l'Amph-B traditionnel, dans des modèles expérimentaux pour l'aspergillose, la candidose et la cryptococcose, bien que les études cliniques chez des volontaires aient été interrompues en raison de son hépatotoxicité. D'autres composés hydrosolubles de ce groupe sont actuellement étudiés.
Nikkomycines.
Les Nikkomycines sont des inhibiteurs de la synthèse de chitine, un composant essentiel des parois cellulaires fongiques.
Nikkomycine Z(Nikkomycin Z, SP-920704, fabricant Shaman) efficace in vivo Et in vitro contre les champignons dimorphes C. immitis Et B. dermatitidis, mais seulement modérément actif in vitro contre C. albicans, Cryptococcus néoformans Et Histoplasma capsulatum. Activité synergique in vitro observé avec l'association de nikkomycine Z avec du fluconazole ou de l'itraconazole contre Candida spp.., Cr. néoformiens Et A. fumigatus Et in vivo- contre H. capsulatum. Accepter per os; synergiste avec le fluconazole et l'itraconazole. La Nikkomycine a été autorisée à Bayer AG en 1995, principalement pour être utilisée dans les mycoses endémiques aux États-Unis, la blastomycose nord-américaine et la coccidioïdose. Nous terminons actuellement des essais précliniques.
Dans ce groupe de médicaments, de nouveaux composés antifongiques (Lys-Nva-FMDP) ont récemment été synthétisés, qui agissent comme un inhibiteur de la glucose-6-phosphate synthétase (une enzyme qui catalyse la première étape de la biosynthèse de la chitine). Suppression de la croissance établie H. capsulatum in vitro Et in vivo, et aucune toxicité lorsqu'il est testé sur des souris.
Une chitinase humaine recombinante a également été créée, qui s'est avérée efficace contre la candidose expérimentale et l'aspergillose chez les animaux, mais a montré une activité significativement plus élevée en combinaison avec l'Amph-B traditionnelle.
Allylamines et thiocarbamates.
Les allylamines et les thiocarbamates sont des agents fongicides synthétiques qui sont des inhibiteurs de l'enzyme squalène époxydase qui, avec la squalène cyclase, convertit le squalène en lanostérol. Dans la paroi fongique, si le squalène n'est pas converti en lanostérol, la conversion du lanostérol en ergostérol est bloquée. En raison de la déplétion en ergostérol, la membrane cellulaire du champignon est endommagée. Il existe deux médicaments antifongiques allylamine, la naftifine et la terbinafine, et un thiocarbamate, le tolnaftate. La naftifine et le tolnaftate sont des médicaments à usage topique, tandis que la terbinafine est utilisée pour le traitement systémique de la dermatomycose.
Terbinafine.
La terbinafine a montré une bonne activité in vitro contre Aspergillus spp., Fusarium spp., dermatomycètes et autres champignons filamenteux, mais activité variable contre les champignons de type levure. Cependant, dans les modèles expérimentaux, il s'est avéré inefficace contre l'aspergillose invasive, la sporotrichose systémique, la candidose systémique ou la cryptococcose pulmonaire. Cependant, une activité a été détectée in vitro contre Aspergillus spp., Candida spp., y compris les souches résistantes aux triazoles, et Pseudallescheria boydii en association avec des azoles ou Amph-B, ainsi que dans des modèles expérimentaux d'aspergillose en association avec Amph-B et dans la sporotrichose cutanée. Actuellement, la terbinafine est principalement utilisée pour le traitement des mycoses cutanées et des onychomycoses, car lorsqu'elle est prise par voie orale, elle crée des concentrations antifongiques dans le lit de l'ongle. La terbinafine est inefficace dans le traitement du pityriasis versicolor car les concentrations qu'elle crée dans la couche cornée ne sont pas élevées pour un effet thérapeutique suffisant. Bien que contrairement à la plupart des azoles, la terbinafine n'inhibe pas le système du cytochrome P450 et en particulier l'isoenzyme CYP3A4, le CYP3A4 peut néanmoins jouer un rôle dans le métabolisme de la terbénafine et ses interactions médicamenteuses. Considérant qu'il est encore métabolisé par d'autres mécanismes hépatiques (uniquement< 5% через систему цитохрома Р450), поэтому некоторые ингибиторы цитохрома Р450 (например, циметидин), могут снижать клиренс тербинафина. Рифампицин увеличивает клиренс фербинафина на 100%. Существует много метаболитов тербинафина, но среди них нет метаболитов с антифунгальной активностью. После приема per os 70 à 80 % de la terbinafine est adsorbée par le tractus gastro-intestinal. Manger n’affecte pas de manière significative sa biodisponibilité, la terbénafine peut donc être prise avec de la nourriture ou à jeun. La terbinafine diffuse rapidement à partir des vaisseaux sanguins (à travers le derme et l'épiderme) et se concentre dans la couche graisseuse. Il est également distribué dans les follicules pileux, les cheveux, la peau, riche en glandes sébacées, restant en concentrations élevées dans les follicules pileux et le lit des ongles. Ses concentrations dans la couche cornée après 12 jours de traitement dépassent les niveaux plasmatiques de 75 fois et dans l'épiderme et le derme de 25 fois. Les cellules sanguines contiennent environ 8 % de la terbinafine administrée ; il manque de sueur. La terbénafine subit un métabolisme de première étape, qui n'implique pas plus de 5 % de la capacité totale du cytochrome P450. Cependant, la terbinafine inhibe de manière compétitive le CYP2D6, ce qui doit être pris en compte lorsqu'elle est utilisée en concomitance avec des médicaments métabolisés par ces isoenzymes (par exemple, l'amitriptyline).
Soldats.
Les soldarines représentent une nouvelle classe d'agents antifongiques potentiels qui inhibent la synthèse des protéines chez les champignons pathogènes. La cible principale de leur action est le facteur d’allongement 2.
De nombreux nouveaux soldarins sont à l'étude, notamment GM-193663, GM-237354, etc. Certains de ces composants ont une activité in vitro contre Candidose spp. , Aspergille spp ., Cryptococcus néoformans, Pneumocysti. carinii et quelques autres champignons. Un effet synergique a été obtenu en combinant les soldarines avec l'Amph-B, l'itraconazole et le voriconazole contre Aspergille spp. et Scedossporium apiospermum. Haute efficacité prouvée in vivo avec candidose et prémonie causées par Pneumocystis carinii. Il est probable que d’autres recherches dans ce domaine se poursuivront.
Peptides cationiques.
Des peptides cationiques d'origine naturelle et artificielle sont incorporés dans les membranes d'ergostérol et de cholestérol de la paroi fongique, ce qui conduit à la lyse cellulaire. Ces peptides ont une activité antifongique contre Aspergille spp. , Candida spp. , Cryptococcus neoformans et Fusarium spp.
Les peptides cationiques naturels comprennent les cécropines, les dermaseptines, l'indolicine, les histatines, le facteur BPI (Augmentation de la perméabilité bactéricide), la lactoferrine et les défensines. Le peptide cationique synthétique Dolastin-10 cible la tubuline intracellulaire et possède une activité fongicide potentielle contre Cr. néoformiens.
Dans ce groupe, Mycoprex (MycoprexD produit par Xoma), obtenu à partir du facteur BPI humain produit par les neutrophiles, fait l'objet d'essais précliniques.
Le choix des médicaments pour diverses mycoses est présenté dans le tableau 7.
Tableau 7.
Médicaments de choix pour diverses infections fongiques.
| Maladie | Traitement |
| Canidose : Candémie Aigu disséminé Chronique disséminée (hépatosplénique) |
Fluconazole |
| Cryptococcose : Pulmonaire Diffusé Avec des dommages au système nerveux central Préventif contre l'infection par le VIH |
Amphotéricine B ou fluconazole Amphotéricine B ou fluconazole Amphotéricine B ou fluconazole Fluconazole |
| Aspergillose | Amphotéricine B standard ou formes liposomales. Itraconazole comme médicament de deuxième intention. |
| Coccidioïdomycose
Sévérité légère à modérée (pulmonaire, disséminée) Lourd |
Fluconazole Amphotéricine B ou fluconazole |
| Blastomycose Pulmonaire Extrapulmonaire Prononcé aigu Méningite |
Itraconazole Itraconazole Amphotéricine B Amphotéricine B |
| Sporotrichose : Ganglions lymphatiques et peau Os et articulations Pulmonaire SNC Prononcé diffusé |
Itraconazole Itraconazole Itraconazole Amphotéricine B Amphotéricine B |
| Trichosporose | fluconazole b amphotéricine b |
| Fusarium | Amphotéricine B régulière ou liposomale |
| Zygomycose ( Mucor spp.) | Amphotéricine B |
| Paracoccidioïdomycose
Sévérité légère à modérée Lourd |
Itraconazole Amphotéricine B |
| Pseudoalleschériose | Kétoconazole ou itraconazole |
(En utilisant les données Andriole V.N., 1999)
Littérature:
- Vanden Bossche H., Marichal P., Odds F. Mécanismes moléculaires de la résistance aux médicaments chez les champignons // Trends Microbiol.-1994.-Vol.2.-P.393-400.
- Hazen E., Brown R. Deux agents anfongiques produits par un actinomycète du sol// Science.-1950.-Vol.112.- P.423.
- Andriole V. T., Kravetz H.M. L'utilisation de l'amphotéricine B chez l'homme // JAMA.-1962.-Vol.180.- P.269-272.
- Georgiev V. S. Traitement et thérapeutique développementale de l'aspergillose//Respiration.-1992.-Vol.59.-P.291-302.
- Aisner J., Schimpff S.C., Wiernik P.H. Traitement de l'aspergillose invasive : relation entre le diagnostic précoce et le traitement et la réponse //Ann.Intern. Med.-1977.-Vol.86.-P.539-543.
- Heidemann H. Th., Gerkens J.F. et coll. Néphrotoxicité de l'amphotéricine B chez l'homme diminuée par la réplétion en sel//Am.J.Med.-1983.-Vol.75.-P.476-481.
- Gonzalez CE, Giri N., Shetty D. et al. Efficacité de la formulation lipidique de nistatine contre l'aspergillose pulmonaire invasive. Dans : Actes et résumés de la 36e conférence Intersciences sur les agents antimicrobiens et la chimiothérapie. Washington, DC : Société américaine de microbiologie, 1996.-Abstr. B54-P.31.
- Karger S. Présentation du SPA-S-843 in vitro activité contre les champignons filamenteux//Chimiothérapie.-2000.-Vol.46.- P.28-35.
- Graybill J.R. Thérapie azolée dans les infections fongiques systémiques. Diagnostic et traitement de l'infection fongique systémique. Raven Press, N-Y., 1989.- P.P.133-144.
- Sucre A. M., Alsip S.G. et coll. Pharmacologie et toxicité du kétoconazole à haute dose // Agents antimicrobiens Chemother.-1987.-Vol.31.- P.11874-1878.
- Chin T., Fong I.W., Vandenbroucke A. Pharmacocinétique du fluconazole dans le sérum et le liquide céphalo-rachidien chez un patient atteint de SIDA et de méningite cryptococcique//Pharmacothérapie.-1990.-V0l.10(4).-P.305-307.
- Ryley J.F.. Chimiothérapie des maladies fongiques. Berlin : Springer-Verlag, 1990.-558 p.
- Edwards D. J. Antifongiques oraux In : Interaction médicamenteuse métabolique (Ed. Revy R.H., Trummel K.E., Trager W.F., Hansen P.D., Eichelbaum A.K.) - Lippincott.Philadelphia, 2000, 793 p.
- Cooker P.J., Tomlinson D.R., Parking J. et coll. Interaction entre le fluconazole et la rifampicine//B.M.J.-1991-Vol.301.- P.818.
- Anaïssie E. J., Kontoyannis D.P. et coll. Concentration plasmatique de sécurité et efficacité du fluconazole à haute dose dans les infections invasives par les moisissures // J. Infect. Dis.-1995.-172-P.599-602.
- Backman J. T., Rivisto K.T., Wang J.-Sh., Neuvonen P.J. Antifongiques In : Interaction médicamenteuse métabolique (Ed. Revy R.H., Trummel K.E., Trager W.F., Hansen P.D., Eichelbaum A.K.) - Lippincott.Philadelphia, 2000.- 793 p.
- Kawakami M., Suzuki K., Ishizuka T. et al. Effet du jus de pamplemousse sur la pharmacocinétique de l'itraconazole chez des sujets sains // Int.J.Clin.Pharmacol.Ther.-1998.-Vol.36.- P.306-308.
- Vanderwoude K., Vodelaers D. et coll. Concentration plasmatique et sécurité de 7 jours d'itraconazole intraveineux suivis de 2 semaines de solution orale d'itraconazole chez les patients en unité de soins intensifs // Antimicrob. Agents Chemother.-1997.-Vol.41.-P.2714-2718.
- Simons K.J., Simons P.E. Antagonistes des récepteurs H1 : pharmacocinétique et pharmacologie clinique. Antagonistes de l'histamine et des récepteurs H1 dans les maladies allergiques Dans : Allergie clinique et immunologie (Ed. M.A.Kaliner).-M.Dekker.-N-Y.-1996.-Vol.7.-P.175-213.
- Lasar J. D., Wilner K.D. Interactions médicamenteuses avec le fluconazole//Rev. Inf. Dis.-1990.-Vol.12, suppl.1.- P.327-333.
- Le juge Como, Dismukes W.E. médicaments azolés oraux comme traitement antifongique systémique // N.Engl.J.Med.-1994.-Vol.330.-263-272.
- Hitchcock C.A., Pye G.W., Oliver G.P. et coll. UK-109, 496, un nouveau dérivé du triazole à large spectre pour le traitement des infections fongiques : activité antifongique et sélectivité in vitro Dans : Actes et résumés de la 35e conférence Intersciences sur les agents antimicrobiens et la chimiothérapie. Washington, DC : Société américaine de microbiologie, 1995.-Absstr. F72.-P.125.
- Denning D., del Favero A., Gluckman E., Norfolk D. et al. UK-109, 496, un nouveau dérivé de triazole à large spectre pour le traitement des infections fongiques : efficacité clinique dans l'aspergillose invasive aiguë // Dans : Actes et résumés de la 35e conférence Intersciences sur l'agent antimicrobien et la chimiothérapie. Washington, DC : Société américaine de microbiologie, 1995.-Abstr. F80.-P.126.
- Sutton D.A., Fothergill A.W., Barchiesi F.J. et coll. In vitro activité du voriconazole contre les champignons dimorphes Dans : Actes et résumés de la 36e conférence Intersciences sur l'agent antimicrobien et la chimiothérapie. Washington, DC : Société américaine de microbiologie, 1996.-Abstr. F85-P.114.
- Radford S.A., Johnson E.M., Warnock D.W. In vitroétudes de l'activité du voriconazole (UK-109 496), un nouvel agent antifongique triazole, contre les agents pathogènes de moisissures émergents et moins courants// Antimicrob. Agents Chemother.-1997.-Vol.41.-P.841-843.
- Purkins L. Voriconazole : Profil pharmacocinétique d'un nouvel azole (Abst. L-23)// Dans : 6ème Congrès de la Confédération européenne de la Société de mycologie médicale.-Barselona, 2000 (Revista de Iberoamericana Micologia.-2000.-Vol.7, f3.-P.114).
- Nomeir A. A., Kumari P., Loebenberg D. et al. Biodisponibilité du SCH56592, un nouvel agent antifingique triazole à large spectre, à partir de diverses formulations Dans : Actes et résumés de la 36e conférence Intersciences sur l'agent antimicrobien et la chimiothérapie. Washington, DC : Société américaine de microbiologie, 1996.-Abstr. F103-P.117.
- Saxon M. Conférence interscientifique sur les agents antimicrobiens et la chimiothérapie -40e réunion (Partie IX) - Toronto, Canada -17-20 septembre 2000.
- Roberts J., Schock K., Marino S., Andriole V.T. Efficacités de deux agents antifongiques, le triazole ravuconazole et l'échinocandine LY-303366, dans un modèle expérimental d'aspergillose invasive//Antimicrob. Agents Chemother.-2000.-Vol.44(12).-P.3381-3388.
- Andriole V.N. Thérapie antifongique actuelle et future : nouvelles cibles pour les agents antifongiques // J. Antimicr. Chemother.- 1999.- Vol.44.- P. 151-162.
- Martinez A., Aviles P., Jiménez E. Activités des soldarins dans des modèles expérimentaux de candidose, d'aspergillose et de pneumocystose // Antimicrob. Agents Chemother.-2000.-Vol.44(12).-P.3389-3394.
Distribution des comprimés Binafin uniquement sur prescription médicale.
Formulaire de décharge
Binafin est produit sous deux formes : crème et comprimés.
1% crème de consistance blanche uniforme et molle. Volume du tube 10, 15, 30 g En application externe, topique.
Les comprimés pour administration orale sont blancs, ronds et biconvexes. Disponible en doses de 125 mg et 250 mg. Le plus souvent, Binafin est disponible à la vente en 14 comprimés, mais il existe également des conditionnements de 10 et 20 comprimés.
Que choisir
Pour le traitement efficace de l'onychomycose, la forme posologique en comprimés est la plus recommandée s'il existe des indications pour un traitement antifongique systémique, mais seul un spécialiste spécialisé peut juger de la nécessité de prescrire des comprimés par voie orale.
Les bienfaits de la crème sont évidents :
- Pour les champignons cutanés, qui accompagnent souvent une infection des ongles ;
- Lorsqu'il est correctement combiné avec d'autres méthodes de traitement. Par exemple, un traitement à la crème de la zone des ongles est nécessaire après une pédicure médicale ;
- L'association avec d'autres formes posologiques (vernis, comprimés) augmentera l'efficacité du traitement de l'onychomycose ;
- À des fins de prévention.
Composition du médicament
Ingrédient actif dans les deux formes de Binafin :
Le chlorhydrate de terbinafine est un composant antifongique à large spectre. Fait face à la plupart des agents pathogènes de l'onychomycose.
Excipients de la crème :
- Ketomacrogol 1000 – émulsifiant, stabilisant pour obtenir une consistance crémeuse ;
- Alcool cétostéarylique – améliore la pénétration des substances, adoucit, crée un film retenant l'humidité à la surface de la peau ;
- Paraffine molle blanche – également connue sous le nom de vaseline. A un effet protecteur, adoucissant et cicatrisant sur les irritations et les plaies. La base pour obtenir une consistance crémeuse ;
- Paraffine liquide - ou huile de vaseline, qui fait partie de la base de la crème ;
- Myristate d'isopropyle – réduit la teneur en matières grasses du mélange, améliore l'application et la répartition de la crème et a un effet adoucissant ;
- Le méthylparabène et le propylparabène sont des conservateurs ;
- Phosphate acide de sodium – affecte la stabilité chimique du médicament ;
- L'eau purifiée est un solvant pour certains composants.
Composants auxiliaires des comprimés :
- Le MCC et l'amidon sont des charges permettant d'obtenir des comprimés ;
- Povidone – se lie aux toxines présentes dans le sang, accélère leur élimination ;
- Le méthylparabène de sodium est un conservateur à activité fongicide prononcée ;
- Laurylsulfate de sodium - destiné à donner la forme d'un comprimé, à conserver sa consistance et sa texture (non dangereux sous cette forme) ;
- Dioxyde de silicium colloïdal – améliore le processus de maintien de la stabilité des comprimés, a un effet positif sur l'absorption de la substance active du médicament ;
- Stéarate de magnésium – charge ;
- Le glycolate d'amidon sodique est une charge qui accélère les processus de décomposition et de biodisponibilité du composant actif ;
- Talc - sert à recouvrir l'enveloppe extérieure du comprimé.
Analogues
Plusieurs dizaines d'analogues contenant de la terbinafine sont produits. Les plus célèbres d'entre eux :
- Lamisil. Disponible sous forme de comprimés, crème, spray. La grande efficacité du médicament original est observée dans la plupart des cas d'utilisation.
- Terbizil, Exiter, Atifin. Ils existent sous les mêmes formes galéniques que Binafin. Préparations de niveau de production européen, avec une efficacité positive testée cliniquement.
- Fungoterbine. Produit national de haute qualité. Contient de l'urée kératolytique et est disponible sous forme de crème et de gel.
Analogues bon marché
L'efficacité et la bonne tolérabilité du médicament Termicon (Pharmstandard) ont été testées par des patients. Il existe des critiques positives sur l'utilisation de Terbinafine-Canonpharma et de Terbinafine (Medisorb).
La crème du même nom Terbinafine est produite par de nombreux fabricants à des prix allant de 60 à 150 roubles. Cependant, l’activité de la substance active n’est souvent pas suffisante pour traiter les infections chroniques. Par conséquent, il est préférable d'utiliser de tels médicaments pour le traitement des mycoses cutanées, mais le résultat évident apparaît plus lentement qu'avec des analogues dont le prix est plus élevé. Des comprimés de Terbinafine bon marché sont également disponibles.
Propriétés du médicament
Le spectre d’action de la terbinafine couvre :
- Champignons dermatophytes :
- Trichophyton (T.rubrum, T.mentagrophytes, T.tonsurans, T.verrucosum, T.violaceum) ;
- Microsporum (M. canis);
- Epidermophyton floccosum.
- Champignons de type levure du genre :
- Candida (Candida albicans);
- Pityrosporum.
- Dimorphique ;
- Moisi.
Une caractéristique de la terbinafine, et donc des médicaments de la gamme Binafin, est son effet fongicide contre les infections, même à de faibles concentrations de la substance. Par rapport aux levures, il peut présenter un effet fongicide ou fongistatique, déterminé par la sensibilité à un certain type de champignon.
Comment ça fonctionne
La substance active du médicament supprime les processus de survie dans la cellule fongique.
Lorsqu'il est utilisé en interne, le médicament s'accumule dans les structures capillaires, les ongles et les tissus cutanés, créant ainsi les conditions d'un effet fongicide constant sur l'infection.
Indications pour l'utilisation
- Infections cutanées dermatophytes ;
- Infections à levures de la peau ;
- Pityriasis versicolor.
Pilules :
- Onychomycose ;
- Mycose du cuir chevelu ;
- Infections cutanées fongiques, y compris les plus courantes.
Mode d'emploi
Mode d'emploi officiel en PDF (131 Ko).
Mode d'application
La crème est appliquée sur les zones péri-unguéales de la peau, car elle pénètre mieux à travers la barrière tissulaire cutanée, mais lors du traitement de la mycose des ongles, les plaques de l'ongle sont également traitées.
- Le produit doit toujours être appliqué sur une peau propre et sèche.
- Une fine couche de crème est légèrement frottée jusqu'à ce que la consistance soit complètement absorbée.
- Un bandage de gaze peut être appliqué sur les espaces entre les doigts pour un effet plus long sur la zone touchée.
Le bord libre de la plaque à ongles infectée par un champignon est coupé au fur et à mesure de sa croissance.
Conditions de stockage
La durée de conservation des comprimés et de la crème est de 3 ans. Conserver dans un endroit sec à une température ne dépassant pas 25 °C. Tenir hors de portée des enfants.
Dosage
Pilules :
La prescription standard pour les adultes est de 1 comprimé 1 fois par jour, 250 mg.
La durée approximative du traitement de l'onychomycose est de 2 à 6 semaines, 6 semaines suffisent pour traiter les ongles, pour les ongles des pieds, la durée peut être de 12 semaines.
La durée du traitement est déterminée individuellement en fonction de la gravité de la maladie. Si la croissance des ongles est lente, le traitement peut être augmenté.
Pour les enfants, les comprimés peuvent être prescrits à partir de 2 ans une fois par jour. Il n'y a aucune information pour une utilisation chez les enfants de moins de 2 ans et pesant moins de 12 kg.
La posologie du médicament est calculée en poids :
- Moins de 20 kg - 62,5 mg (1/4 comprimé de 250 mg ou 1/2 comprimé de 125 mg) ;
- De 20 kg à 40 kg - 125 mg (1 comprimé de 125 mg ou 1/2 comprimé de 250 mg) ;
- Plus de 40 kg - 250 mg (1 comprimé de 250 mg).
Il n'y a aucune raison d'ajuster la dose des comprimés pour les personnes âgées ; vous devez être attentif aux contre-indications en général.

Appliquer 1 à 2 fois par jour.
La durée du traitement des mycoses cutanées est d'environ 1 à 2 semaines. Le produit doit être utilisé même après les premières améliorations visibles, ce qui évitera la récidive de l'infection.
La thérapie complexe de l'onychomycose nécessite une utilisation plus longue de la crème et peut durer 1 mois ou plus.
La forme crème est contre-indiquée pour les enfants de moins de 12 ans.
Aucun ajustement posologique n’est nécessaire chez les patients âgés.
Surdosage
Des pilules. Avec l'utilisation interne de Binafin, des maux de tête, des étourdissements, des nausées et des douleurs dans l'épigastre sont possibles. Le traitement consiste en un lavage gastrique et la prise de médicaments entérosorbants.
Crème. Aucun cas de surdosage n'a été constaté pour un usage externe.
Caractéristiques d'utilisation avec d'autres médicaments
Crème. Il n'y a aucune preuve d'interactions médicamenteuses.
Des pilules. Des études menées chez des volontaires sains ont montré une faible capacité à augmenter ou diminuer la clairance des médicaments impliqués dans le métabolisme du système du cytochrome P450 (cyclosporine, terfénadine, tolbutamide, triazolam et contraceptifs oraux).
L'élimination de la terbinafine est ralentie par les médicaments qui inhibent le cytochrome P450 (cimétidine) et accélérée par les médicaments qui augmentent le taux de métabolisme sanguin (rifampicine).
La prudence est de mise lors de l'utilisation simultanée de médicaments des groupes suivants :
- Antidépresseurs tricycliques sélectifs ;
- Inhibiteur sélectif de la recapture de la sérotonine;
- Inhibiteurs de la MAO de type B ;
- Bêta-bloquants.

Schémas d'application
La pratique du traitement de l'onychomycose prouve la grande efficacité de la thérapie combinée. Par conséquent, l’utilisation combinée de comprimés Binafin et d’un traitement local avec la crème Binafin accélère le début de la récupération.
Si l'utilisation de Binafin n'est pas associée à Binafin de la forme posologique opposée, mais à un analogue, le pronostic du traitement reste le même. Par exemple, comprimés Lamisil + crème Binafin ou comprimés Binafin + crème Atifin.
Contre-indications
- Hypersensibilité individuelle à la composition ;
- Grossesse.
Sous la surveillance d'un médecin
Lors de la prescription de comprimés aux femmes pendant l'allaitement, l'allaitement est arrêté car la terbinafine est excrétée dans le lait maternel.
Effets secondaires
Crème. Très rarement, des rougeurs, des démangeaisons et des brûlures sont possibles au site d'application. En cas d'effets indésirables, le médicament est arrêté.
Des pilules. En général, ils sont bien tolérés. Tous les effets secondaires disparaissent après l'arrêt du médicament.
Une prudence particulière et une prescription appropriée sont nécessaires en cas de maladies chroniques du foie et des reins.
Dans de rares cas, il est possible :
- Troubles digestifs (perte d'appétit, dyspepsie, diarrhée) ;
- Allergies cutanées ;
- Effet négatif sur les muscles musculaires (arthralgie, myalgie).
Si les effets secondaires persistent pendant une longue période, Binafin est arrêté.
Commentaires
Avantages et inconvénients du produit
- Coût relativement faible par rapport au chlorhydrate de terbinafine original ;
- Tolérance satisfaisante ;
- Efficace contre divers agents pathogènes de la mycose des ongles ;
- Bonne absorption de la crème.
- Possibilité d'une efficacité moindre par rapport au médicament d'origine.
Un examen par un médecin de l'analogue le plus proche de la binafine - la terbinafine - sera utile, car les recommandations et les actions des médicaments sont courantes :
Terbinafine contre les champignons : mode d'emploi
Caractéristiques et composition du médicament

La pommade Terbinafine est bien absorbée par la peau. Pénétrant dans les couches profondes de l'épiderme, il affecte la membrane cellulaire de la cellule fongique, qui meurt ensuite. La concentration maximale du médicament est observée deux heures après l'application ou l'ingestion sous forme de comprimé. La terbinofine n'est pas retenue dans l'organisme et est excrétée après 24 heures dans les urines. La pommade Terbinafine a un effet positif sur la peau affectée par les champignons et l'herpès, grâce aux composants inclus dans la composition. Le principal ingrédient actif, la terbinafine, est complété par les composants suivants :
- eau purifiée;
- l'alcool benzylique;
- glycérol distillé;
- la vaseline;
- acide stéarique;
- émulsifiant n°1 ;
- triéthanolamine.
Les substances actives contenues dans le comprimé de Terbinafrine peuvent réagir avec des médicaments contenant du cytochrome P450 (contraceptifs oraux, cyclosporine, tolbutamide), et Terbinafrine Teva est également incompatible avec les bloqueurs de l'histamine H2.
La crème Terbinafine est active dans la lutte contre presque toutes les souches du champignon, donc si vous ne constatez pas d'amélioration, vous devez refaire un diagnostic. Il est probable qu’une autre maladie non liée à une infection fongique ait pu provoquer les symptômes.
Indications pour l'utilisation

La terbinafine est utilisée le plus souvent contre la mycose des ongles, mais en plus d'éliminer rapidement cette maladie, la pommade contre le lichen et l'herpès de toute localisation a montré son efficacité. Le médicament a également les indications d'utilisation suivantes :
- mycose;
- pied d'athlète;
- trichophytose;
- candidose;
- rubrophytie;
- microsporose.
En plus des effets positifs, l'utilisation du médicament peut également être nocive, surtout s'il existe l'une des contre-indications :
- insuffisance rénale;
- dysfonctionnement hépatique;
- conditions pathologiques des vaisseaux sanguins;
- violation du métabolisme des protéines et des glucides dans le corps;
- manque de lactase dans le corps;
- inhibition de la fonction hématopoïétique ;
- néoplasmes tumoraux;
- enfance;
- intolérance individuelle au médicament ou hypersensibilité du corps.
La terbinafine ne doit pas être prise pendant la grossesse ; il existe une possibilité que les composants inclus dans la composition perturbent le développement de l'embryon et provoquent une fausse couche. En outre, le médicament peut réduire la qualité du lait et n'est donc pas utilisé pendant l'allaitement. Une contre-indication à l'utilisation du médicament est que la date de péremption est expirée et que l'intégrité de l'emballage est compromise.
Pour les enfants atteints de mycose des ongles, d'autres médicaments ou analogues de Terbinafine Teva sont utilisés, car l'effet de la substance active elle-même n'a pas encore été entièrement étudié sur un organisme en croissance. Je voudrais également noter que Terbinafrine Teva n'aide pas s'il y a de l'alcool dans le corps ; pendant le traitement, vous devez absolument renoncer aux boissons alcoolisées.
Méthode d'utilisation

La pommade fongique Terbinafine s'utilise en externe 2 fois par jour. Le médicament est appliqué sur l'ongle dans la zone d'infections fongiques ou sur la zone cutanée où sont notés une éruption herpétique et des symptômes de lichen. La durée et la posologie de la crème Terbinafine pour la mycose des ongles peuvent varier ; le régime est ajusté par le médecin traitant en fonction de l'état du patient et de la présence de maladies supplémentaires.
La pommade Terbinafine mff peut être utilisée sous forme de compresses en cas de candidose cutanée étendue. Le produit est appliqué en couche dense sur un tissu en coton et appliqué sur la zone affectée. Changez le pansement une fois par jour. La durée d'un tel traitement avec un agent antifongique est de 14 jours, selon les critiques, ce temps est largement suffisant pour éliminer l'infection.
De nombreux patients s'intéressent à ce qui est meilleur, crème ou pommade, en fait, c'est assez difficile à dire. L'usine pharmaceutique de Moscou produit trois formes pour usage externe de ce médicament et une pour usage interne. Le gel contient la concentration la plus élevée de substance active, il est donc recommandé de l'utiliser en présence d'ulcères suintants et en cas de libération d'exsudat. La crème est appliquée lorsque la plaie est légèrement sèche et la pommade est utilisée lorsque la plaie est sèche.
Les champignons des ongles et de la peau peuvent être éliminés en prenant des comprimés, comme dans le cas d'une pommade ; il est interdit d'utiliser le médicament pour traiter un enfant. Il est conseillé aux adultes de prendre 1 comprimé deux fois par jour pendant une semaine. Vous pouvez prendre le médicament uniquement avec de l'eau non gazeuse afin que le médicament soit mieux absorbé dans l'estomac.
Si une personne ignore la liste des contre-indications ou ajuste la posologie à sa discrétion, elle peut ressentir des effets secondaires. Lorsqu'il est appliqué à l'extérieur, l'état pathologique se manifestera par une éruption rouge, un gonflement de la zone touchée par le champignon, ainsi que de fortes démangeaisons. Si le médicament est pris par voie orale, des étourdissements, des nausées, des vomissements et une miction accrue peuvent survenir.
Pour devis : Comparaison du traitement continu par la terbinafine avec la thérapie pulsée par l'itraconazole pour l'onychomycose des pieds // RMJ. 2001. N° 11. P. 482
Pour le traitement systémique de l'onychomycose, on utilise actuellement la terbinafine (Lamisil) du groupe des allylamines, qui a un effet fongicide, et l'itraconazole (Orungal), qui appartient aux triazoles et a un effet fongistatique. Le traitement par la terbinafine est généralement effectué en continu pendant 12 semaines, l'itraconazole est utilisé en continu (en même temps) ou pendant 1 semaine par mois pendant 3 à 4 mois (thérapie par impulsions), et un certain nombre d'auteurs considèrent la thérapie par impulsions comme efficace au fur et à mesure d'un traitement continu par l'itraconazole ou la terbinafine. Une étude prospective multicentrique, randomisée en double aveugle, a examiné l'efficacité et la sécurité du traitement continu par la terbinafine par rapport à la thérapie pulsée par l'itraconazole chez les patients atteints d'onychomycose des pieds. L'étude, menée sur 72 semaines, a impliqué 35 centres dans 6 pays européens. Le groupe d'étude était composé de 496 patients âgés de 18 à 75 ans atteints d'onychomycose des pieds cliniquement et mycologiquement confirmée, causée par des dermatophytes. Les patients ont été randomisés en 4 groupes parallèles et ont reçu un traitement par terbinafine 250 mg/jour pendant 12 (groupe T12) ou 16 semaines (T16) ou par itraconazole 400 mg/jour (4 gélules de 100 mg) pendant 1 semaine de chaque mois pendant 3 ou 4 mois (groupes I3 et I4).
Le principal critère de jugement était le taux de guérison mycologique, déterminé par des résultats microscopiques et de culture négatifs du matériel prélevé sur les ongles cibles (gros ongle). Les taux de guérison mycologique ont été évalués après 72 semaines de suivi. Les critères d'efficacité secondaires comprenaient le taux de guérison clinique (clairance de l'ongle à 100 %), le taux de guérison complète (mycologique et clinique), l'efficacité clinique (guérison mycologique, croissance d'au moins 5 mm d'un nouvel ongle non affecté par une mycose) et l'évaluation globale. donné par le médecin et le patient.
Les agents responsables étaient : Trychophyton rubrum (89,3%), T.mentagrophytes (8,5%), T.rubrum+ moisissures non dermatophytes (1,6%), T.rubrum + T.mentagrophytes (0,6%).
Le taux de guérison mycologique après 72 semaines était de : 75,7 % et 80,8 % dans les groupes T12 et T16, 38,3 % et 49,1 % dans les groupes I3 et I4 (voir figure). Le taux de guérison clinique était significativement plus élevé avec tous les schémas thérapeutiques à la terbinafine qu'avec le traitement pulsé par l'itraconazole (p<0,0022). На протяжении всего исследования (вплоть до 72-й недели) частота микологического и клинического излечения в обеих группах тербинафина продолжала повышаться, тогда как в группах итраконазола она не менялась.
Riz. Taux de guérison mycologique (%)
Le taux de guérison complète et l'efficacité clinique étaient significativement plus élevés dans les groupes terbinafine que dans les groupes itraconazole (p<0,005). Общая оценка результатов лечения выявила достоверное преимущество непрерывного лечения тербинафином по сравнению с пульс-терапией итраконазолом (р<0,0001).
236 patients ont signalé des événements indésirables (55, 61, 60 et 60 dans les groupes T16, T12, I3, I4, respectivement). Les caractéristiques des événements indésirables pour les 4 groupes ne différaient pas de manière significative et se situaient dans les profils d'innocuité connus des deux médicaments.
Les résultats du traitement ont été jugés bons ou très bons par 79 à 85 % des médecins et des patients des groupes terbinafine et seulement 44 à 55 % des groupes itraconazole.
L'étude a montré qu'un traitement par terbinafine 250 mg/jour pendant 12 ou 16 semaines permet d'obtenir un taux de guérison mycologique et clinique plus élevé par rapport à la thérapie pulsée par itraconazole après 72 semaines de suivi.
Une explication possible de l’efficacité plus élevée de la terbinafine dans cette étude réside dans les différences de concentrations fongicides et fongistatiques des deux médicaments décrites dans la littérature. La terbinafine a un effet fongicide contre les dermatophytes et sa concentration fongicide minimale (MFC) est d'environ 0,004 µg/ml. L'itraconazole a un effet fongistatique et son MPA moyen contre les dermatophytes est d'environ 0,6 µg/ml. Lors du traitement à la terbinafine, des concentrations de médicament dans l'ongle sont créées 100 fois supérieures à son MPA, tandis que lors de l'utilisation d'itraconazole, la concentration de médicament dans l'ongle se situe uniquement à la frontière entre les concentrations fongistatiques et fongicides. Les fluctuations des concentrations d'itraconazole observées chez différents patients peuvent aggraver les résultats du traitement, tandis que l'utilisation de la terbinafine, malgré des fluctuations importantes des concentrations, assure la destruction de l'agent pathogène. L’avantage thérapeutique de la terbinafine a été plus clairement démontré dans cette étude, puisqu’elle incluait des patients atteints d’onychomycose sévère avec des lésions relativement étendues et une évolution longue de la maladie.
Classification:
Polyènes – nystatine
Azoles – flucosanol, kétoconazole
Allylamines – terbinafine
Azolés :
Spectre d'activité : Les principaux agents responsables des candidoses (albicans, tropicales), les dermatomycètes.
NDR : Dyspepsie du système nerveux central (maux de tête, vertiges, paresthésies, tremblements, convulsions), réactions allergiques (éruption cutanée, démangeaisons), hépatotoxicité (augmentation de l'ALT, de l'AST, ictère)
Allylamines :
Dermatomycètes, candida, aspergillus.
NDR : identique aux azoles
Médicaments anti-inflammatoires de base. Classification, effets, NDR, indications d'utilisation.
(toujours à la question 25)
La base du traitement de la PR et du LED est constituée d'AINS + médicament de base. Les médicaments de base, par rapport aux AINS, suppriment le processus inflammatoire plus profondément, mais l'effet thérapeutique se développe plus lentement (semaines, mois).
Méthotrexate
L’étalon-or pour la RA. Antagoniste de l'acide folique. A un effet immunosuppresseur prononcé même à petites doses
Composés d'or
Perturbe l'activation des lymphocytes T et le développement d'une réaction auto-immune
NDR : démangeaisons, dermatite, protéinurie, diarrhée. En cas de complications importantes - dimercaprol (un médicament qui lie l'or)
Pénicillamine :
Significativement inférieur à l’or en termes d’efficacité et de tolérance
NDR : Syndrome néphrotique, ictère, myasthénie grave.
Sulfasalazine
Tolérance supérieure à la pénicillamine
NDR : nausées, vomissements, éruption cutanée
Chloroquine :
La tolérance est bonne, mais beaucoup moins efficace que les autres
NDR : rarement – dermatite, myopathie
Principes de choix d'un médicament antimicrobien, voie d'administration et schéma posologique.
La thérapie antimicrobienne est de deux types : étiotrope et empirique. Étiotropique - lorsque l'agent pathogène est connu, empirique - lorsqu'il n'est pas connu. Il est souvent nécessaire de recourir à une thérapie empirique, car... l'identification de l'agent pathogène prend plusieurs jours, et parfois même n'est pas possible du tout. Pour augmenter l'efficacité de la thérapie empirique, certains principes doivent être suivis :
1. Le choix du médicament doit être basé sur un diagnostic précis, cela vous permet de déterminer au moins l'agent pathogène suspecté. 2. La préférence est donnée aux médicaments ayant un spectre d'action plus étroit. 3. Ne prescrivez pas de médicaments antibactériens pour une infection virale.
Voies d'administration
Il faut considérer :
1. S'il est pris par voie orale, est-il absorbé depuis les intestins vers le sang ?
2. S'ils pénètrent dans les tissus mous, sont-ils libérés des muscles et pénètrent dans la circulation sanguine ou provoquent-ils la destruction des tissus mous.
3. Est-il possible d’injecter le médicament dans la circulation sanguine ?
4. Ou il est préférable d'utiliser le médicament localement par inhalation.
Schéma posologique
Lors du choix des doses, il faut tenir compte de la capacité des médicaments antimicrobiens à pénétrer une barrière particulière. Par exemple, la bezylpénicilline. Il peut endommager le cortex cérébral, mais il est incapable de pénétrer la barrière hémato-encéphalique. Il appartient donc à une large dose AB. La dose, par exemple, de céphalosporines peut également varier, mais pas plus de 5 fois. Ils appartiennent aux AB à dosage limité. Eh bien, par exemple, les macrolides, les aminosides, leurs doses ne peuvent différer de plus de 2 fois. Ils concernent des AB strictement dosés. Ce sont les plus dangereux, car la différence entre la concentration permettant d'obtenir un effet thérapeutique et le maximum admissible n'est pas grande.
Principes d'antibiothérapie rationnelle, résistance aux antibiotiques.
AB doit être prescrit en fonction de la sensibilité d'un agent pathogène particulier à celui-ci
AB doit être prescrit à une dose et administré de manière à assurer une concentration thérapeutique au site de l'inflammation.
AB doit être prescrit à une dose et administré de manière à limiter autant que possible ses effets néfastes.
Résistance aux antibiotiques
Résistance primaire – associée aux caractéristiques génétiques de l’espèce
Secondaire – se produit pendant le traitement de l’AB
Critères d'évaluation de l'efficacité et de la sécurité de la thérapie antibactérienne. Exemples.
Efficacité:
Normalisation de t, disparition des symptômes. Diminution du nombre de leucocytes, ESR, CRP.
Sécurité:
Pour évaluer la sécurité des AB, il est nécessaire de détecter d’éventuels effets indésirables en clinique et en laboratoire. Par exemple, lors de la prise de médicaments ayant des effets néphrotoxiques, surveillez la fonction rénale (créatinine dans le sang).