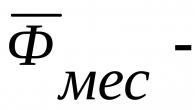7379 0
Les patients présentant un déficit en lymphocytes T sont sensibles aux infections virales, fongiques et protozoaires. De plus, comme les lymphocytes T sont nécessaires pour que les lymphocytes B produisent des anticorps contre les antigènes T-dépendants, les patients présentant un déficit en lymphocytes T présentent également des défauts sélectifs dans la production d'anticorps. Par conséquent, les patients présentant un déficit en lymphocytes T peuvent être difficiles à distinguer des patients atteints de SCID sur la base de la présentation clinique.
Aplasie thymique congénitale (syndrome de DiGeorge)
syndrome de DiGeorge est un déficit en lymphocytes T dans lequel le thymus, ainsi que d'autres organes non lymphoïdes, se développent anormalement. Ce syndrome est dû à une migration altérée des cellules de la crête neurale embryonnaire dans les troisième et quatrième poches pharyngées. Normalement, ce processus se produit à la 12e semaine de gestation. Dans le syndrome de DiGeorge, des anomalies du cœur, de la région faciale, du thymus et des glandes parathyroïdes se développent, conduisant à une aplasie thymique et à une hypoparathyroïdie. La conséquence de l’aplasie thymique est l’absence de cellules T matures et un déficit immunitaire. Le modèle du syndrome de DiGeorge est constitué de souris nues ; ces animaux ne développent pas de thymus ni de follicules pileux.Le syndrome de DiGeorge n'est pas héréditaire, mais survient sporadiquement à la suite d'une délétion du chromosome 22q11. Les nouveau-nés souffrent d'hypocalcémie (faibles niveaux de calcium) en raison de l'absence de glandes parathyroïdes et de malformations cardiaques congénitales. Ces enfants souffrent d’infections virales, bactériennes, fongiques et protozoaires fréquentes ou chroniques. Ils ne possèdent pas ou très peu de lymphocytes T matures en périphérie (dans le sang, les ganglions lymphatiques ou la rate). Bien que les taux de lymphocytes B, de plasmocytes et d’Ig plasmatiques puissent être normaux, de nombreux patients ne présentent pas d’augmentation de la réponse en anticorps après une immunisation avec des antigènes T-dépendants.
Le manque de lymphocytes T auxiliaires nécessaires pour passer à d’autres isotypes d’immunoglobulines entraîne l’absence d’IgG et d’anticorps d’autres isotypes après la vaccination. Cependant, la réponse IgM aux antigènes T-indépendants n’est pas affectée. Étant donné que les patients atteints du syndrome de DiGeorge ont un déficit en lymphocytes T et ne sont pas capables de générer une réponse normale en anticorps, ils ne peuvent pas être vaccinés avec des vaccins viraux vivants atténués !
Initialement, les enfants atteints du syndrome de DiGeorge ont été traités par transplantation de thymus fœtal, ce qui a entraîné l'apparition de lymphocytes T chez l'hôte en une semaine. Le thymus fœtal utilisé pour la transplantation doit dater de moins de 14 semaines de gestation pour garantir l'absence de GVHD, qui peut survenir si des cellules de thymus matures d'un donneur sont administrées à un receveur immunodéprimé.
Le thymus fœtal du donneur fournit des cellules épithéliales thymiques qui permettent aux cellules progénitrices lymphoïdes normales du receveur de se développer en lymphocytes T. Bien que les cellules T résultantes soient normales, l’immunité à médiation cellulaire et l’aide à la production d’anticorps ne sont pas entièrement restaurées. Les cellules T du receveur « se souviennent » des molécules du CMH des cellules thymiques transplantées comme « les leurs » et interagissent parfois mal avec les cellules présentatrices d'antigènes de leur propre corps en périphérie.
Cette stratégie thérapeutique n’ayant pas donné de résultats, la thérapie symptomatique est désormais principalement utilisée. Certains patients présentent du tissu thymique résiduel, qui assure, bien que retardé et réduit, le développement des cellules T. Un problème médical supplémentaire associé à ce syndrome est la cardiopathie congénitale, qui aggrave un pronostic autrement sombre.
Défaillance des lymphocytes T avec un nombre normal de lymphocytes T périphériques
Chez un certain nombre de patients, les anomalies des lymphocytes T se sont avérées fonctionnelles plutôt que quantitatives. Cliniquement, ils peuvent présenter des infections opportunistes et une incidence étonnamment élevée de maladies auto-immunes. Une étude des pedigrees dans ces familles révèle un mode de transmission autosomique récessif. Les études moléculaires démontrent l'hétérogénéité des facteurs responsables, y compris la sous-expression de la tyrosine kinase ZAP-70, CD3ε ou CD3ε (Fig. 17.4).Riz. 17.4. Déficit en molécules impliquées dans la transmission du signal via le récepteur spécifique de l'antigène (récepteur des lymphocytes T)
ZAP-70 est requis pour la transduction du signal intracellulaire lors de la liaison au récepteur des lymphocytes T. Pour des raisons peu claires, les patients présentant une expression défectueuse de ZAP-70 présentent un tableau clinique d'un syndrome de type SCID (avec des défauts de l'immunité à médiation cellulaire et humorale). Le manque d’activité des lymphocytes T suggère que ZAP-70 joue un rôle essentiel dans la fonction des lymphocytes T matures, mais les raisons de son effet sur la fonction des lymphocytes B ne sont pas claires. De plus, bien que le nombre de cellules sanguines périphériques, les ganglions lymphatiques et le thymus soient initialement normaux, les patients présentant une expression défectueuse de ZAP-70 manquent de lymphocytes T CD8+.
Cette découverte suggère que ZAP-70 est également nécessaire à la différenciation des cellules T CD8+ dans le thymus. Les mutations des chaînes CD3 sont assez rares et un petit nombre de patients présentant ce défaut ont été décrits. Les modèles animaux suggèrent que les chaînes peptidiques CD3 sont nécessaires à la signalisation normale des récepteurs des lymphocytes T. Cependant, il n’est pas clair si ces modèles de souris correspondent exactement au défaut présent chez les patients décrits précédemment.
Syndrome lymphoprolifératif auto-immun
Syndrome lymphoprolifératif auto-immun (ALPS)- une maladie héréditaire caractérisée par une prolifération massive de tissu lymphoïde avec développement précoce d'un lymphome. Elle peut être considérée comme auto-immune car une telle anomalie génétique entraîne un phénomène auto-immun systémique et une susceptibilité accrue uniquement aux infections virales chroniques. Les patients présentent un nombre accru de lymphocytes T double-négatifs (CD4CD8) et peuvent développer un lymphome à cellules B. La plupart des patients atteints d'ALPS présentent une mutation du gène codant pour la protéine Fas (CD95).Les signaux transmis par cette protéine activent normalement l’apoptose, ou mort cellulaire programmée. Sans activation de l’apoptose, les cellules qui devraient mourir vivent et les réponses immunitaires qui devraient être « désactivées » se poursuivent. La plupart des patients atteints du syndrome lymphoprolifératif auto-immun ont une molécule Fas normale et une molécule Fas mutante. Cela suggère que la molécule mutante interfère d’une manière ou d’une autre avec le fonctionnement de la molécule Fas normale. Certains patients atteints du syndrome lymphoprolifératif auto-immun présentent des défauts dans d'autres composants déclenchant l'apoptose, tels que le ligand Fas ou la caspase 10.
Deux races de souris - lps et gld - ont un phénotype similaire à celui des patients atteints d'ALPS ; Les souris lps ont une mutation dans le gène Fas et les souris gld ont une mutation dans le gène du ligand Fas. Pendant de nombreuses années, les souris lps ont été étudiées comme modèle de maladie auto-immune, en particulier le LED, jusqu'à ce qu'un défaut dans leur gène Fas soit découvert.
Candidose cutanéo-muqueuse chronique
Candidose cutanéo-muqueuse chronique est un ensemble mal défini de syndromes caractérisés par une infection de la peau et des muqueuses par des champignons du genre Candida. Ces champignons sont généralement présents à la surface de ces tissus mais ne sont pas pathogènes. Les patients ont généralement une immunité à médiation cellulaire intacte contre les organismes autres que Candida et une immunité à médiation cellulaire normale (production d'anticorps) contre tous les organismes, y compris Candida. Ainsi, il n’y a qu’un défaut sélectif dans la fonction des lymphocytes T. Cette maladie touche aussi bien les hommes que les femmes, est particulièrement fréquente dans l'enfance et peut dans certains cas être héréditaire.Maladies d'immunodéficience associées aux cellules B ou aux immunoglobulines
Les maladies immunitaires associées aux cellules B ou aux immunoglobulines vont des maladies avec un développement défectueux des cellules B et une absence totale de toutes les classes d'Ig aux maladies associées au déficit d'une classe ou d'une sous-classe d'Ig. Les patients souffrent d'infections récurrentes ou chroniques qui peuvent débuter dès l'enfance (agammaglobulinémie de Bruton) ou au début de l'âge adulte. Pour diagnostiquer ces maladies, il est nécessaire de connaître le nombre de lymphocytes B et d'étudier leur fonction, de procéder à une détermination immunoélectrophorétique et quantitative des classes et sous-classes d'Ig.Agammaglobulinémie infantile liée à l'X
Agammaglobulinémie infantile liée à l'X (XIA) décrit pour la première fois en 1952 par O. Bruton. Cette maladie est également appelée agammaglobulinémie de Bruton. Cette maladie est relativement rare (1 cas pour 100 000 personnes). Elle apparaît pour la première fois entre 5 et 6 mois, lorsque le bébé perd les IgG maternelles reçues par le placenta. À cet âge, chez les nourrissons, les principales manifestations sont des infections bactériennes sévères et récurrentes résultant d'une suppression sévère ou éventuellement d'une absence d'immunoglobulines de toutes classes.Le principal défaut est l’incapacité des précurseurs des lymphocytes B, présents en nombre normal, à se développer en lymphocytes B matures. Le gène BTK (Brutonian tyrosine kinase), qui subit des mutations dans le CSA, code normalement pour une enzyme tyrosine kinase située dans le cytosol. Il semble être un participant essentiel au processus de transduction du signal provenant du récepteur des cellules pré-B dans les cellules B en développement. Sans ce signal, le développement cellulaire ne se poursuit pas.
Toutes les cellules B matures chez les individus ayant reçu le gène mutant de leur mère se forment uniquement en raison de la présence d'un autre chromosome X actif (non mutant). Ainsi, la CSA est l'une des nombreuses immunodéficiences héréditaires décrites pour lesquelles une mutation de la tyrosine kinase cytoplasmique est responsable. Des mutations dans JAK3, qui provoquent le développement d'une forme de SCID, et dans ZAP-70, qui sont responsables d'une forme de défaillance des lymphocytes T, ont été décrites précédemment.
Les études du sang, de la moelle osseuse, de la rate et des ganglions lymphatiques des patients atteints d'ASC révèlent une absence presque totale de cellules B et de plasmocytes, ce qui explique les faibles taux d'Ig. Il est courant que les enfants aient des amygdales considérablement sous-développées. Les lymphocytes B, produits en nombre limité, ont la capacité normale de devenir des plasmocytes. Les nourrissons atteints d'ASC souffrent d'otites moyennes bactériennes récurrentes, de bronchite, de septicémie, de pneumonie, d'arthrite, de méningite et de dermatite. Les micro-organismes les plus fréquemment détectés sont Hemophilus influenzae et Streptococcus pneumoniae.
Souvent, les patients souffrent également de malabsorption en raison de la prédominance de Giardia lamblia dans le tractus gastro-intestinal. De plus, ces patients sont également sensibles aux infections virales qui pénètrent dans le tractus gastro-intestinal, telles que les échovirus et le poliovirus. Les infections ne répondent pas bien aux antibiotiques seuls, le traitement consiste donc en des injections périodiques immunoglobuline intraveineuse (IVIG), contenant une grande quantité d’IgG. Même si cette immunisation passive soutient certains patients pendant 20 à 30 ans, le pronostic est prudent car la probabilité de développer des maladies pulmonaires chroniques est élevée en raison d'infections récurrentes fréquentes.
Hypogammaglobulinémie transitoire
Chez les nourrissons âgés de 5 à 6 mois, les IgG maternelles transférées passivement disparaissent et la production d'immunoglobulines dans le corps commence à augmenter. Les nourrissons prématurés peuvent présenter un déficit transitoire en IgG car ils ne sont pas encore capables de synthétiser des immunoglobulines. Parfois, les nourrissons nés à terme peuvent également présenter une diminution des taux d'IgG, même avec des taux d'IgM et d'IgA normaux. Cette condition est due à une diminution du nombre de cellules T auxiliaires et à une perturbation de leur fonction.L'hypogammaglobulinémie transitoire peut persister de plusieurs mois à 2 ans. Cette maladie n’est pas associée au sexe et se distingue d’une maladie liée à l’X par la présence d’un nombre normal de lymphocytes B dans le sang. Bien qu'un traitement ne soit généralement pas nécessaire, ces nourrissons doivent être identifiés car la vaccination ne doit pas être administrée pendant cette période.
Déficit immunitaire variable courant
Les patients présentant un déficit immunitaire commun variable (DICV) ont des taux sériques d'IgG et d'IgA considérablement réduits, avec des taux d'IgM normaux ou réduits et un nombre normal ou réduit de lymphocytes B dans le sang périphérique. La cause de cette maladie, qui touche aussi bien les hommes que les femmes, n’est pas entièrement comprise et peut ne pas être la même dans tous les cas. L'apparition de la maladie peut survenir à tout âge, mais il y a deux pics entre 1 et 5 ans et entre 15 et 20 ans. Les personnes touchées souffrent d'infections respiratoires et intestinales récurrentes causées par des bactéries pyogènes et de maladies auto-immunes telles que l'anémie hémolytique, la thrombocytopénie et le LED, qui sont associées à des auto-anticorps. Beaucoup souffrent également de troubles de l’immunité à médiation cellulaire. À long terme, ces patients présentent une incidence accrue de cancers, en particulier de lymphomes et de cancers gastriques.Le déficit immunitaire variable courant est caractérisé par une maturation altérée des cellules B en cellules sécrétant des anticorps. Ce défaut peut être dû à l'incapacité des cellules B à proliférer en réponse à l'antigène, ou à une prolifération normale des cellules B sans sécrétion d'IgM, ou à une sécrétion d'IgM sans commutation isotopique en IgG ou IgA (en raison de dommages intrinsèques aux cellules B ou T), ou à une altération. glycosylation des chaînes lourdes d'IgG. Dans la plupart des cas, le trouble se manifeste par une inhibition de la synthèse et de la sécrétion des immunoglobulines. Cette maladie est familiale ou sporadique et les facteurs environnementaux à l’origine de la maladie sont inconnus.
Le traitement dépend de la gravité. Dans les formes sévères de la maladie avec de nombreuses infections répétées ou chroniques, un traitement par immunoglobuline intraveineuse est prescrit. Les patients traités ont généralement une espérance de vie normale. Les mères atteintes de CVID ont des grossesses normales, mais elles ne transmettent certainement pas les IgG maternelles au fœtus.
Déficits sélectifs en immunoglobulines
Plusieurs syndromes sont provoqués par des déficits sélectifs d’une classe ou d’une sous-classe d’immunoglobulines. Certains d'entre eux s'accompagnent d'une augmentation compensatoire des taux d'autres isotypes d'anticorps, comme par exemple une augmentation de la concentration d'IgM en cas de déficit en IgG ou en IgA.Le déficit en IgA est le trouble d’immunodéficience le plus répandu en Occident, avec une incidence estimée à une personne sur 800. La cause de ce trouble est inconnue mais semble être liée à une diminution de la libération d'IgA par les cellules B. Un déficit en IgA peut également survenir de manière transitoire, en tant qu’effet secondaire des médicaments. Les patients peuvent souffrir d'infections récurrentes des voies respiratoires supérieures et inférieures d'étiologie virale ou bactérienne, de la maladie coeliaque (altération de l'absorption intestinale) ou ne présenter aucun symptôme.
Le traitement des patients qui développent des manifestations de la maladie consiste à prescrire des antibiotiques à large spectre. Le traitement par immunoglobulines sériques n'est pas utilisé car les préparations commerciales contiennent de faibles concentrations d'IgA et parce que les IgA administrées par voie parentérale n'atteignent pas les sites du système immunitaire sécrétoire où elles constituent normalement une immunoglobuline protectrice. De plus, les patients peuvent développer une réponse humorale (généralement des IgG ou des IgE) aux IgA provenant du sérum immun transfusé, provoquant des réactions d'hypersensibilité. Cependant, le pronostic du déficit sélectif en IgA est généralement bon et la plupart des patients vivent bien.
Des déficits sélectifs d'autres isotypes d'immunoglobulines sont également notés. Un exemple est le déficit en IgM, une maladie rare dans laquelle les patients souffrent d'infections répétées et graves causées par des micro-organismes dotés de capsules de polysaccharides, tels que les pneumocoques ou Haemophilus influenzae. Des déficits sélectifs en sous-classes d’IgG ont été décrits mais sont extrêmement rares.
Troubles de l'interaction entre les lymphocytes T-B
Il existe au moins deux maladies dans lesquelles les lignées de cellules T et B mûrissent normalement, mais les interactions entre leurs membres sont perturbées. Bien que ces deux affections soient causées par des anomalies des lymphocytes T, les manifestations cliniques prédominantes sont liées aux lymphocytes B ou à la réponse immunitaire humorale. Ces maladies sont le syndrome hyper-IgM et la maladie lymphoproliférative liée à l'X.Syndrome d'hyper-IgM
Syndrome hyper-IgM lié à l'X (CHIM) se manifeste à l'âge de 1 à 2 ans par des infections respiratoires récurrentes et des taux sériques très faibles d'IgG, d'IgA et d'IgE en association avec des taux d'IgM normaux ou élevés (voir Fig. 17.1, défaut 8). Le nombre de lymphocytes B chez ces patients est normal ; in vitro, ils sont pleinement fonctionnels et capables de changer d'isotype avec une stimulation appropriée. Les lymphocytes T sont également présents en nombre normal, normalement répartis entre les sous-populations et répondent par prolifération aux mitogènes. Une mutation du gène CD40L4 situé sur le chromosome X entraîne l'absence de ligand CD40 (CD154) sur les cellules Tn.Dans ce cas, le CD40L se lie au CD40 exprimé sur les cellules B. Cette interaction empêche l’apoptose des lymphocytes B et semble être importante, voire essentielle, pour le changement d’isotype. Avec le CGIM, il n’y a pas non plus d’attraction des cellules B vers les follicules, ce qui conduit à l’absence de centres germinaux formés. Les garçons atteints de ce trouble présentent également des changements subtils dans la fonction des lymphocytes T et un blocage partiel de la différenciation des neutrophiles et de l'activation des macrophages. Cela peut expliquer leur susceptibilité aux infections opportunistes, en particulier la pneumonie à Pneumocystis carinii, et leur pronostic est pire que celui des patients atteints d'ASC.
Le deuxième groupe de patients présentant les mêmes manifestations cliniques que dans le CGIM, mais avec une transmission de type autosomique récessive, peut présenter un défaut des lymphocytes B dans la molécule CD40. Chez les patients du troisième groupe présentant un défaut dans l'interaction du CD40 avec le modulateur du facteur de transcription NF-κB, un mécanisme d'hérédité lié à l'X est observé. Comme c'est souvent le cas avec les troubles génétiques impliquant ces types de molécules régulatrices des interactions intercellulaires, les enfants affectés subissent également des changements pathologiques dans des cellules qui n'appartiennent pas au système immunitaire. Par exemple, ces molécules régulatrices utilisent de nombreux types de cellules.
Trouble lymphoprolifératif lié à l'X (syndrome de Duncan)
D'abord Lymphoprolifératif lié à l'X (XL) la maladie a été décrite chez six parents masculins de la famille Duncan, le nom est donc devenu un nom familier. La principale composante du défaut complexe qui détermine le développement de cette maladie rare est l’incapacité des cellules T à réguler la croissance des cellules B. Avant l’exposition au virus d’Epstein-Barr, ces patients sont cliniquement sains et possèdent un nombre normal de lymphocytes T et B. Cependant, lorsqu’ils sont exposés au virus, ils développent une forme grave de mononucléose infectieuse, qui peut être mortelle. Si le patient survit à l’infection, il développe souvent un lymphome malin ou une dysgammaglobulinémie.Un lymphome ou un déficit en immunoglobulines peuvent survenir chez eux sans contact préalable avec le virus d'Epstein-Barr. Parmi les lymphomes, les lymphomes agressifs à cellules B sont majoritairement observés en dehors des ganglions lymphatiques (extraganglionnaires), en particulier dans le tractus gastro-intestinal. Le type le plus courant est le lymphome de Burkitt. Bien que le type de lymphomes qui se développent soit le même que chez les patients présentant d'autres troubles du contrôle de la prolifération des lymphocytes B induite par le virus d'Epstein-Barr et provoqués par un lymphocyte T défectueux (par exemple, les patients atteints du SIDA ou les patients recevant un traitement immunosuppresseur après une transplantation), le L'incidence du développement de lymphomes dans le CSL est significativement plus élevée. Le pronostic est extrêmement défavorable.
Dysfonctionnement des cellules phagocytaires
Les cellules phagocytaires - leucocytes polymorphonucléaires et macrophages/monocytes - jouent un rôle très important dans l'immunité innée et acquise, agissant contre les agents pathogènes seuls ou en association avec les lymphocytes. Les syndromes de déficit héréditaire des cellules phagocytaires ont permis d'identifier une variété de molécules nécessaires à toutes les étapes des actions phagocytaires nécessaires pour tuer l'agent pathogène.
Riz. 17.5. (A) Une adhésion cellulaire altérée altère la capacité des globules blancs à interagir avec l’endothélium vasculaire, provoquant une perturbation lorsque ces cellules migrent de la circulation sanguine vers le site de l’infection. (B) Les défaillances des mécanismes nécessaires à la phagocytose entraînent une perturbation de la destruction intracellulaire des micro-organismes
Ces étapes et leurs déficits associés comprennent la migration et l'adhésion des cellules phagocytaires (déficit d'adhésion des leucocytes), la phagocytose et la fusion avec les lysosomes (syndrome de Chediak-Higashi) et l'explosion oxydative pour tuer l'agent pathogène (maladie granulomateuse chronique) (Fig. 17.5). Le dysfonctionnement phagocytaire peut également être secondaire, provoqué par des facteurs externes tels que des médicaments et des maladies systémiques (par exemple le diabète), ou par des défauts dans d’autres parties du système immunitaire.
Déficit d’adhésion leucocytaire
Pour que les globules blancs arrivent au site d’infection des tissus, ils doivent d’abord quitter la circulation sanguine. Ce processus comprend plusieurs étapes. Dans un premier temps, les leucocytes commencent à rouler lentement le long de l'endothélium en raison des interactions des sélectines sur les cellules endothéliales et des ligands de la sélectine sur les leucocytes. Puis, sous l’influence de chimioattractants, la cellule cesse de bouger (rouler). Une telle cellule s'attache plus étroitement, puis commence à migrer à travers l'endothélium. La dernière étape nécessite l'interaction des intégrines sur les leucocytes avec leurs ligands sur les cellules endothéliales.Déficit d’adhésion leucocytaire (LAD)- il s'agit d'un groupe de troubles dans lesquels l'interaction des leucocytes avec l'endothélium vasculaire est interrompue (Fig. 17.5, A). NLA I est une maladie autosomique récessive dont le gène est cartographié sur le chromosome 21. Les patients reçoivent un diagnostic de défaut dans la sous-unité β des molécules d'intégrine, ce qui empêche leur expression. De plus, la sous-unité β est commune à trois intégrines exprimées sur les granulocytes, les monocytes et les lymphocytes - LFA-1 (CD1 la/CD18), Mac-1 (CD1 lb/CD18) et p150.95 (CD1 lc/CD18), respectivement. . En conséquence, l’adhésion et la migration de tous types de leucocytes sont perturbées.
Les patients atteints de NPA I souffrent d'infections bactériennes récurrentes des tissus mous dans lesquelles leur nombre de globules blancs augmente mais ne produit pas de pus ou ne guérit pas efficacement. Les fonctions lymphocytaires sont également altérées en raison du manque d’expression du LFA-1. Une caractéristique distinctive des nouveau-nés atteints de NLA I est la perte tardive du reste du cordon ombilical.
Les patients atteints de NLA II présentent un défaut dans les ligands de la sélectine, de sorte que leurs cellules ne peuvent pas rouler le long de la surface endothéliale (la première étape de la migration) (voir Fig. 17.5, A). Le défaut qui détermine le développement de NLA II est associé au métabolisme du fucose, ce qui conduit à l'absence de ligands fucosylés auxquels pourraient se lier les sélectines. Les symptômes d'immunodéficience dans ce trouble sont moins prononcés. En plus d'eux, un défaut du métabolisme du fucose entraîne d'autres défauts de développement. Comme pour le NPA I, l'absence ou la formation insignifiante de pus est caractéristique ; Ces enfants ne présentent pas de signes cliniques classiques d’inflammation lors d’infections graves.
Syndrome de Chédiak-Higashi
Le syndrome de Chediak-Higashi est une maladie autosomique récessive caractérisée par des granules géants et des organites anormaux dans les cellules (Fig. 17.5, B). Les lysosomes et les mélanosomes sont principalement touchés, entraînant des défauts de pigmentation et un dysfonctionnement des neutrophiles, des cellules NK et des plaquettes, ainsi que des anomalies neurologiques. Les neutrophiles ont une capacité réduite à tuer des organismes au niveau intracellulaire, ce qui est le résultat à la fois d'une dégranulation défectueuse et d'une fusion altérée des lysosomes avec les phagosomes. Au fil du temps, les patients développent des infiltrats massifs de lymphocytes et de macrophages dans le foie, la rate et les ganglions lymphatiques. Les micro-organismes pyogènes, tels que les streptocoques et les staphylocoques, provoquent des infections récurrentes, parfois mortelles. Le pronostic est défavorable.Maladie granulomateuse chronique
À maladie granulomateuse chronique (CGD) la dernière étape de destruction des organismes absorbés est perturbée (voir Fig. 17.5, B). Dans ce cas, la persistance intracellulaire des organismes conduit à la formation de granulomes. Chez les individus normaux, les neutrophiles activés et les phagocytes mononucléaires détruisent les micro-organismes par une explosion oxydative qui consomme de l'oxygène et libère du peroxyde d'hydrogène et des radicaux libres superoxydes. Des mutations dans l’une des sous-unités de l’enzyme qui catalyse cette explosion, la NADPH oxydase, peuvent conduire à la CGD.La forme la plus courante de CGD est due à une mutation dans l'une des sous-unités associées à la membrane, gp91phox, codée par le gène SUVV situé sur le chromosome X. Ainsi, la plupart des patients ont un mode de transmission récessif lié à l'X. D'autres sous-unités de la NADP oxydase sont codées par des gènes autosomiques. Les patients atteints de CGD causée par des mutations dans de telles sous-unités autosomiques présentent un mode de transmission autosomique récessif. Ils présentent principalement des mutations dans l’une des deux sous-unités cytosoliques de l’enzyme, p47phox ou p67phox.
Les symptômes de la maladie apparaissent au cours des 2 premières années de la vie. Les patients présentent une susceptibilité accrue à l'infection par des micro-organismes qui ont normalement une faible virulence, tels que Staphylococcus aureus, Serratia marcescens et Aspergillus.
Les anomalies associées comprennent la lymphadénopathie (hypertrophie des ganglions lymphatiques) et l'hépatosplénomégalie (hypertrophie du foie et de la rate), associées à des infections chroniques et aiguës. Le traitement consiste en une immunisation agressive et un traitement avec des antibiotiques à large spectre, des antifongiques et de l'interféron-γ.
En plus du CGD, les troubles dans lesquels il existe une diminution ou une absence d'activité des enzymes impliquées dans le processus de phagocytose entraînent une diminution de la capacité à détruire les organismes intracellulaires. Ceux-ci comprennent la glucose-6-phosphate déshydrogénase, la myéloperoxydase et la phosphatase alcaline.
Déficit en récepteur d'interféron-γ
Une mutation du gène IFNγR1 entraîne l’incapacité des monocytes à répondre à l’IFNγ en sécrétant le facteur de nécrose tumorale a (TNFα). Les patients porteurs de cette mutation sont sélectivement sensibles aux mycobactéries légèrement pathogènes, confirmant l'importance de l'IFNy dans le contrôle des infections mycobactériennes. Cette sélectivité suggère également que d'autres activités de l'IFNy sont compensées chez les individus atteints de ce trouble. La vaccination par le BCG, courante dans certaines régions du monde, peut être dangereuse pour les patients présentant cette anomalie.Déficit en cellules NK
On sait très peu de choses sur le déficit en cellules tueuses naturelles chez l'homme ; Seuls quelques cas de cette maladie ont été décrits. Des études animales suggèrent que le déficit en cellules NK altère le rejet de l'allogreffe et est associé à une susceptibilité accrue aux maladies virales et à une incidence accrue de métastases tumorales. Des anomalies des cellules NK sont observées dans les troubles d'immunodéficience combinés sévères, certains troubles de la fonction des cellules T et des cellules phagocytaires et le syndrome lymphoprolifératif lié à l'X.Maladies causées par des troubles du système complémentaire
Le complément est nécessaire pour l'opsonisation et la destruction des bactéries et des cellules endommagées, la chimiotaxie et l'activation des cellules B. Les composants du complément sont également impliqués dans la destruction des complexes antigène-anticorps, empêchant ainsi le dépôt de complexes immuns et les maladies ultérieures. La carence en complément peut être héritée comme un trait autosomique, les individus hétérozygotes ayant la moitié de la concentration normale d'un composant particulier du complément. Pour la plupart des composants, cela suffit à prévenir les manifestations cliniques de la maladie. La demi-vie des composants activés du complément est également généralement strictement contrôlée par des inhibiteurs qui détruisent les produits d'activation ou dissocient les complexes.Déficit en complément en phase d’activation précoce
Les composants du complément en phase d'activation précoce sont particulièrement importants pour la formation de l'opsonine C3b (Fig. 17.6). Chez les patients présentant un déficit en C1, 4 ou 2 le long de la voie classique d'activation ou un déficit en C3 lui-même, les cas d'infection par des organismes encapsulés (Streptococcus pneumoniae, Streptococcus pyogenes, Haemophilus influenzae) et de maladies rhumatismales dues à une clairance altérée des complexes immuns sont plus fréquents. en raison d'une formation insuffisante de C3b. La conséquence la plus notable est la survenue fréquente de maladies auto-immunes. En effet, le LED est l’une des manifestations les plus courantes de carence en certains composants du complément.Chez ces patients, le LED apparaît très précocement et est plus grave que le LED non associé à un déficit en complément. De plus, un tel LED peut se développer en l’absence d’anticorps souvent retrouvés dans d’autres cas. La carence en lectine liant le mannose, qui s'attache à la surface des microbes sans la participation d'anticorps et déclenche la voie classique d'activation du complément, entraîne également un risque accru d'infections bactériennes et de symptômes de type lupus. Étant donné que toutes les voies d'activation du complément - classique, lectine et alternative - nécessitent l'activation du C3, le déficit en C3 lui-même est associé aux symptômes les plus graves, en particulier à l'évolution compliquée des maladies infectieuses.
Carence en complément en phase d’activation tardive
En cas d'insuffisance des composants du complément de la phase d'activation tardive (C5 - C9), la formation du complexe d'attaque membranaire (MAC) est perturbée. Ce complexe est un destructeur direct des bactéries à Gram négatif, en particulier Neisseria meningitidis, et constitue la première ligne de défense contre elles (voir Fig. 17.6).Contrôle altéré des composants du complément
Angio-œdème héréditaire. Dans les cas d'angio-œdème héréditaire, les patients ne disposent pas d'un inhibiteur efficace de la C1-estérase. Si cet inhibiteur est absent, l'action de C1 sur C4, C2 et le système kallicréine est incontrôlée, entraînant la formation de grandes quantités de peptides vasoactifs.
Riz. 17.6. La cascade d'activation du complément montre que lorsque les composants du complément en phase d'activation précoce sont déficients, les patients sont prédisposés aux infections causées par des micro-organismes encapsulés et aux syndromes rhumatoïdes. Un déficit en complément en phase d'activation tardive est associé aux infections à Neisseria
Ces peptides provoquent une augmentation de la perméabilité des vaisseaux sanguins. Les patients présentent un gonflement localisé, qui peut mettre la vie en danger s'il se développe dans le larynx, obstruant les voies respiratoires. Le traitement consiste à éliminer les facteurs déclenchants, qui incluent généralement une blessure, et à administrer des perfusions d'un inhibiteur de la C1 estérase.
Carence en protéines associée au phosphatidylinositol glycosylé. La famille de protéines ancrées au GPI est exprimée sur les membranes des globules rouges, des lymphocytes, des granulocytes, des cellules endothéliales (cellules qui tapissent les vaisseaux sanguins) et des cellules épithéliales. Ces protéines, qui comprennent le facteur accélérateur de dissociation (CD55) et CD59, protègent les cellules de la lyse spontanée par le complément. Si les inhibiteurs sont absents à la surface cellulaire, les granulocytes, les plaquettes et surtout les érythrocytes sont susceptibles d'être lysés spontanément par le complément. Il n'existe que quelques familles présentant des mutations héréditaires dans les protéines FUD, CD59 ou toutes les protéines GPI. Ces patients présentent des symptômes d'anémie sévère, de thrombose et d'infections chroniques.
La forme acquise de cette maladie, appelée hémoglobinurie paroxystique nocturne (HPN), se produit beaucoup plus souvent. Dans l’HPN, les patients ne disposent pas de l’enzyme nécessaire pour produire toutes les protéines ancrées au GPI. Cela se produit en raison d’une mutation somatique acquise dans une cellule progénitrice myéloïde précoce. Trois lignées non lymphoïdes sont touchées : les granulocytes, les plaquettes et les érythrocytes.
Chez la plupart des patients, les cellules souches présentant cette mutation acquièrent des mutations supplémentaires, puis commencent à dominer les cellules normales de la moelle osseuse et arrêtent de mûrir, conduisant à une leucémie myéloïde aiguë. Au cours de l'évolution chronique de l'HPN, une hémolyse intravasculaire se développe, particulièrement visible la nuit dans les reins, où l'environnement acide déclenche l'activation du complément le long de la voie alternative. Cette manifestation clinique se reflète dans le nom de la maladie.
Maladies d'immunodéficience secondaire
Les déficits immunitaires secondaires sont des conséquences d’autres maladies. La mauvaise nutrition (ou son absence) reste la cause la plus fréquente de troubles d’immunodéficience dans le monde. Dans les pays développés, l'immunodéficience iatrogène est plus fréquente, c'est-à-dire causées par un traitement médicamenteux, notamment du fait de l'utilisation d'agents chimiothérapeutiques dans le traitement du cancer ou d'une immunosuppression contrôlée en cas de transplantation d'organes ou de maladies auto-immunes.Des déficits immunitaires secondaires se développent également en cas de maladies auto-immunes (et non à la suite d'un traitement) ou après la guérison d'infections bactériennes graves. Les tumeurs malignes du système immunitaire suppriment également souvent les composants normaux (non malins), ce qui entraîne une susceptibilité accrue aux maladies infectieuses chez ces patients.
R. Koiko, D. Sunshine, E. Benjamin
Le rôle du thymus, principal organe du système immunitaire, n'a été déterminé qu'en 1961 grâce aux données de R. A. Good et J. F. Miller. Malgré la disponibilité de nombreuses données sur le rôle du thymus dans la maturation du système lymphoïde, de nombreux aspects de ce problème, notamment la transformation de cellules lymphoïdes préthymiques immatures en cellules T immunocompétentes présentant une hétérogénéité fonctionnelle importante, n'ont pas été entièrement élucidés. .
Les lymphocytes T qui ont traversé le thymus acquièrent la capacité de répondre aux antigènes et mitogènes (PHA et ConA), s'installent dans les zones thymus-dépendantes des organes lymphoïdes (dans les zones paracorticales des ganglions lymphatiques, follicules péri-artériels de la rate) et constituent la majeure partie du pool de recirculation dans le sang périphérique avec un cycle de vie long. Ces cellules sont dites thymo-dépendantes.
Développement des lymphocytes T
Le développement intrathymique est assuré par différents facteurs : un « microenvironnement » spécifique déterminé par le stroma du thymus, ainsi que ses hormones, ses cellules épithéliales et mésenchymateuses. Un certain nombre de substances actives sont isolées du thymus, différant par leur activité, leur poids moléculaire et leur résistance aux influences thermiques. Parmi eux, la thymosine, la thymopoïétine et le facteur thymique humoral sont décrits. Ces substances, notamment la thymosine, favorisent la transformation des lymphocytes T vers des formes plus matures.
Le mécanisme d'action de la thymosine et d'autres substances actives sur les cellules T n'est pas tout à fait clair ; on pense que leur point d'application est la membrane cellulaire, à savoir le système adénylate cyclase. Probablement, la thymosine l'active et augmente la concentration d'AMPc et de GMPc intracellulaires. Ces nucléotides sont liés aux processus de développement cellulaire. Ces derniers sont très complexes, diversifiés et nécessitent une étude plus approfondie.
Types et fonctions des cellules T
Parmi les lymphocytes T, on distingue plusieurs sous-populations, qui diffèrent par leur sensibilité au cortisol et par leurs différences au niveau des antigènes et des récepteurs à leur surface.
De par leur objectif, les lymphocytes T sont principalement responsables de l'immunité cellulaire, mais dans leurs fonctions, ils sont hétérogènes. Ils se composent de plusieurs sous-classes : les cellules qui reconnaissent l'antigène ; cellules auxiliaires; cellules tueuses ; cellules suppressives; cellules mémoire, etc. La question de savoir quand les lymphocytes T sont divisés en sous-populations ayant des objectifs fonctionnels différents est très complexe et controversée. On ne sait pas clairement ce qui détermine l’orientation fonctionnelle du développement, si ces sous-populations proviennent d’un seul précurseur ou si la différence est génétiquement déterminée au niveau de la cellule souche.
Tous les sous-ensembles de lymphocytes T représentent environ 60 à 70 % de tous les lymphocytes ; environ 25 % sont des cellules B, ou cellules dépendantes de la bourse, qui ont subi un « entraînement » dans un autre organe central de l'immunité - la bourse de Fabricius (chez les oiseaux) ou ses analogues chez l'homme, où elles acquièrent des propriétés distinctes des cellules T. Il convient de noter qu’une longue recherche de cet analogue n’a pas abouti. Ce rôle est attribué à diverses formations lymphoïdes - l'appendice, les amygdales, les groupes de follicules lymphatiques des intestins, les accumulations lymphoïdes dans les poumons et d'autres organes. Après maturation, les lymphocytes B se déplacent principalement vers les follicules spléniques et les ganglions lymphatiques.
La plupart des antigènes dépendent du thymus, car la réponse immunitaire à ceux-ci nécessite la participation des lymphocytes T ; ces antigènes sont complexes, possèdent plusieurs déterminants antigéniques avec des spécificités différentes (tissulaires, microbiens, viraux).
Le mécanisme de reconnaissance antigénique par les cellules T est très complexe et n’a pas été entièrement étudié. La tâche principale des cellules T et de leurs récepteurs est de reconnaître le « soi » et le « non-soi ». antigènes. L’étiologie des récepteurs des lymphocytes T qui reconnaissent les antigènes n’a pas été définitivement établie. L'interaction des récepteurs cellulaires avec l'antigène est un signal pour le déclenchement d'une réponse immunitaire et d'une différenciation cellulaire. Si la réponse immunitaire suit le type de formation d’anticorps, alors la participation des lymphocytes B et leur transformation en plasmocytes nécessite la participation des lymphocytes T auxiliaires (Th). Le développement des réactions cellulaires réalisées par les lymphocytes T se produit également avec la participation d'assistants - les amplificateurs T (Ta).
Le mécanisme de l'action auxiliaire des lymphocytes T (contact direct ou substances actives sécrétées par les cellules) est très complexe et n'est pas entièrement compris. Les Th activés produisent des facteurs spécifiques et activent les cellules B via les macrophages. L'essence de ces facteurs n'a pas été entièrement déterminée. Étant donné que les lymphocytes B produisent différentes classes d’immunoglobulines, il est possible que différentes sous-populations Th existent pour réguler la formation d’anticorps (IgG, IgE).
Gershon a développé en 1969 le concept du rôle suppressif des cellules T (suppresseurs T), puis il a été découvert que les T sont capables de réguler diverses phases de la réponse immunitaire. Le rôle des suppresseurs est de réguler le niveau et la force d'une réponse immunitaire spécifique, garantissant une tolérance immunologique à certains antigènes thymus-dépendants et aux antigènes de leurs tissus. Les Ts portent des récepteurs pour les IgG, les IgM et agissent par l'intermédiaire de médiateurs solubles. La nature de ces facteurs sécrétés par Ts est activement étudiée. Il n’existe pas de consensus sur les mécanismes d’induction de Ts (contact direct des cellules avec l’antigène ou signaux de rétroaction).
La valeur de Ts est très élevée ; limiter la réaction immunitaire n'est pas moins important pour l'organisme que la stimuler. Cela s'applique particulièrement aux réactions immunitaires pouvant conduire à une pathologie (maladies auto-immunes, maladies)
Les lymphocytes T tueurs réalisent des réactions immunitaires cellulaires : effets cytopathogènes sur l’immunité de transplantation, l’immunité antitumorale et certains types d’infections. Une sous-population de cellules tueuses est également un effecteur d’hypersensibilité de type retardé (DTH).
L'hétérogénéité de la population de lymphocytes T en termes de propriétés fonctionnelles explique l'interaction extrêmement complexe des cellules dans les réactions de l'organisme aux antigènes.
Il est désormais évident que non seulement les lymphocytes T, mais aussi les lymphocytes B ne constituent pas non plus une masse homogène de cellules. Les lymphocytes B se caractérisent par la présence à la surface d'immunoglobulines, qui assurent la reconnaissance d'antigènes, sous l'influence desquels les lymphocytes B se transforment en plasmocytes produisant des anticorps -. Leur production pour la plupart des antigènes ne peut se produire sans interaction avec les cellules T. La coopération cellulaire est une condition nécessaire au développement d’une réponse immunitaire.
Les lymphocytes T activés, en plus des facteurs sécrétés par Ts et Th, forment un certain nombre de médiateurs - les lymphokines, qui ont des propriétés physico-chimiques différentes. Leur rôle est l'implication de diverses cellules dans la réponse immunitaire - granulocytes neutrophiles, macrophages, granulocytes éosinophiles. La participation de ces lymphocytes T à la réponse immunitaire est très importante. Le rôle des macrophages, qui détruisent l'antigène et le transforment en une forme immunogène, est particulièrement important. Les macrophages comprennent actuellement un groupe de cellules capables de phagocytose, adhérant aux verres - adhérents, cellules mononucléées phagocytaires, macrophages tissulaires et monocytes.
L'article a été préparé et édité par : chirurgienLeucémie à cellules T de l'adulte
Le virus de la leucémie à cellules T de l'adulte est endémique, c'est-à-dire qu'il est distribué parmi les humains uniquement dans certaines régions du monde. La majeure partie se trouve au Japon et dans les îles des Caraïbes. Des niveaux élevés d'infection ont été constatés parmi les aborigènes de Papouasie-Nouvelle-Guinée, d'Australie et des Îles Salomon. Un autre foyer de ce virus s’est formé autour de la mer Caspienne. En Russie, le virus n'est étonnamment présent que parmi les Nivkhs du village de Nogliki, situé dans la partie centrale de l'île de Sakhaline. Le virus se transmet de personne à personne par l’allaitement, les rapports sexuels et la transfusion de sang contaminé (ou lorsque des toxicomanes partagent une seringue).
Dans les zones d’endémie, de nombreuses personnes sont infectées par le virus, mais, en règle générale, les personnes infectées deviennent porteuses asymptomatiques du virus à vie. Seulement chez 2 à 3 % des porteurs, après une longue période de latence de plusieurs décennies, le virus brise son « vœu de silence ». Dans ce cas, une maladie maligne se développe, dans laquelle le nombre de lymphocytes immatures augmente fortement, le foie et la rate grossissent, le tissu osseux est détruit et une éruption cutanée se développe souvent.
La cible du virus de la leucémie à cellules T sont les lymphocytes T. Après l’infection, le virus insère son matériel génétique dans le chromosome de l’hôte. Bien que le virus ne possède pas son propre oncogène, les protéines virales activent un grand nombre de gènes cellulaires, notamment des oncogènes cellulaires. Ainsi, les cellules infectées dans lesquelles le virus est soudainement activé deviennent malignes et commencent à se multiplier de manière incontrôlable. De plus, ils améliorent le travail des gènes qui dirigent la synthèse des interleukines - de petites protéines à l'aide desquelles les cellules du système immunitaire communiquent entre elles. Une forte augmentation de la quantité d'interleukines, provoquée par le virus de la leucémie à cellules T, crée un bruit d'information qui désorganise le fonctionnement du système immunitaire. En particulier, le nombre de lymphocytes T tueurs diminue. Le système immunitaire est privé du moyen le plus important pour vaincre les cellules tumorales et n’est plus en mesure de faire face à leur expansion. Par conséquent, le pronostic de cette maladie est sombre : l’espérance de vie ne dépasse généralement pas six mois après le diagnostic.
Le virus de la leucémie à cellules T adultes est l’un des plus anciens compagnons de l’homme. On pense que son origine remonte à environ 20 000 ans. On l'a trouvé chez les Indiens d'Amérique du Sud et chez les pygmées africains, c'est-à-dire chez les représentants de tribus longtemps isolées du monde extérieur. L’étude de la diversité génétique de ce virus permet de retracer les routes migratoires des anciens humains. En particulier, une comparaison des isolats asiatiques et américains du virus de la leucémie à cellules T a fourni une preuve supplémentaire de l'hypothèse selon laquelle les ancêtres des Indiens d'Amérique étaient des Mongoloïdes d'origine asiatique. Il y a probablement 10 à 40 000 ans, ils ont pénétré en Amérique le long de l'isthme qui reliait alors l'Asie et le Nord.
L'Amérique à la place de l'actuel détroit de Béring, et s'installe sur tout le continent américain.
Extrait du livre Grande Encyclopédie Soviétique (CL) de l'auteur BST Extrait du livre Grande Encyclopédie Soviétique (LE) de l'auteur BST Extrait du livre Grande Encyclopédie Soviétique (SHK) de l'auteur BST Extrait du livre Encyclopédie de l'étiquette d'Emily Post. Règles de savoir-vivre et savoir-vivre raffinés pour toutes les occasions. [Étiquette] par le message de Peggy Extrait du livre Biologie [Ouvert de référence complet pour la préparation à l'examen d'État unifié] auteur Lerner Georgy IsaakovichANNIVERSAIRES ADULTES Les conjoints, les enfants et parfois les amis proches du garçon d'anniversaire souhaitent célébrer solennellement l'une de ses dates « rondes », comme le 30e, le 40e ou le 50e anniversaire. Il n'y a pas de règles ou de restrictions particulières en la matière. Célébrer un tel événement
Extrait du livre Meubles en osier par Antonov E.1.3. Les principaux niveaux d'organisation de la nature vivante : cellulaire, organisme, population-espèce, biogéocénotique Termes et concepts de base testés dans les épreuves d'examen : niveau de vie, systèmes biologiques étudiés à ce niveau,
Extrait du livre Guide complet de diagnostic médical auteur Viatkina P. Extrait du livre Atlas : anatomie et physiologie humaines. Guide pratique complet auteur Zigalova Elena YurievnaLeucémie Une faiblesse, une léthargie et un malaise sont observés chez les patients atteints de leucémie aiguë et chronique. Les leucémies aiguës comprennent les maladies tumorales du système sanguin dont le substrat principal sont les cellules blastiques : myéoloblastes, lymphoblastes, monoblastes, érythroblastes,
Extrait du livre Encyclopédie complète d'une jeune femme au foyer auteur Polivalina Lyubov AlexandrovnaLeucémie Pour les leucémies aiguës myéloblastiques, lymphoblastiques et toutes les autres formes de leucémie aiguë chez l'adulte, le régime VAMP est efficace ( cure de 8 jours : méthotrexate - 20 mg/m2 par voie intraveineuse les jours 1 et 4, vincristine - 2 mg/m2 par jour les jours 2 ). jour 1 du traitement intraveineux, 6-mercaptopurine -
Extrait du livre Encyclopédie des méthodes de développement précoce auteur Rapoport Anna Extrait du livre Fauteuils, chaises, tables, étagères et autres meubles en osier auteur Podolski Youri FedorovitchJeux pour adultes Des vacances ne sont pas des vacances si elles se transforment en une banale beuverie. Et pour éviter que cela n'arrive, nous vous conseillons, hôtesses, de ne pas oublier les jeux et divertissements qui peuvent se jouer directement à table « Sortez la réponse » C'est très simple et en même temps.
Extrait du livre Armoires coulissantes, couloirs, toboggans, murs, étagères, commodes et autres meubles préfabriqués auteur Podolski Youri Fedorovitch Extrait du livre Encyclopédie du Dr Myasnikov sur les choses les plus importantes auteur Miasnikov Alexandre Léonidovitch Extrait du livre Le guide le plus complet de l'éleveur de volailles auteur Slutsky Igor Extrait du livre de l'auteur5.4. Vaccinations pour adultes Tout adulte, en particulier les femmes en âge de procréer, doit s'assurer d'avoir été vacciné contre les oreillons, la rubéole et la rougeole. Si à l’époque soviétique la rougeole était réduite au minimum, nous en récoltons aujourd’hui les conséquences.
Le lymphome à cellules T appartient à un groupe de pathologies cancéreuses non hodgkiniennes qui affectent le système lymphatique. Cette maladie hématologique touche principalement les hommes âgés, mais peut survenir chez les femmes. La maladie est d'origine épidernotrope et se caractérise par un développement agressif.
Classification des maladies non hodgkiniennes
Le système lymphatique humain constitue la principale défense humaine contre les maladies infectieuses. Les principaux assistants dans la lutte contre les virus sont les lymphocytes.
Ces éléments sanguins sont divisés en trois types :
Les maladies non hodgkiniennes surviennent en raison de modifications de ces cellules, capables de muter et de se multiplier rapidement. Selon le nom des leucocytes impliqués dans le développement de la maladie, les lymphomes sont divisés en tumeurs à cellules NK, B et T.
Parmi les néoplasmes non hodgkiniens à cellules B, la pathologie la plus courante est :
- lymphome à cellules du manteau;
- tumeur folliculaire;
- plasmocytome;
- néoplasme dans la zone marginale;
- lymphome à petites cellules.
Les tumeurs à cellules NK sont formées à partir de leucocytes NK atypiques. Les néoplasmes provoqués par des mutations des lymphocytes T comprennent :
Ce sont des maladies de structure et de structure différentes, caractérisées par la prolifération de cellules malignes dans les tissus lymphoréticulaires. Le processus pathologique couvre la moelle osseuse, les ganglions lymphatiques, le système digestif, le foie et la rate. Les tumeurs non hodgkiniennes sont beaucoup plus fréquentes que la maladie de Hodgkin.
Caractéristiques des lymphomes à cellules T
Selon l'évolution de la maladie, on distingue les lymphomes non hodgkiniens indolents et agressifs. La maladie des faux tissus est également extrêmement rare. Cette maladie est similaire aux tumeurs cancéreuses, mais est un néoplasme de nature hématologique.
Les pathologies indolentes sont divisées en plusieurs sous-types :

Les tumeurs indolentes sont des néoplasmes passifs à développement lent. Les lymphomes agressifs diffèrent par l'intensité de leur croissance. Cette maladie comprend :
- syndrome de Sézary ;
- lymphome cutané périphérique agressif et primitif à cellules T ;
- le lymphome est préliminaire ;
- tumeur extraganglionnaire;
- leucémie de l'adulte.
Parfois, les tumeurs passives peuvent évoluer en lymphomes avec une évolution agressive de la maladie. Dans d’autres cas, la pathologie des lymphocytes T se développe à un rythme moyen.
Causes de pathologie
Les raisons de ces changements dans le système lymphatique n'ont pas été entièrement identifiées. Les scientifiques pensent que le principal facteur dans la formation de tumeurs malignes des leucocytes T est le virus des cellules T leucémiques humaines de type 1. Fondamentalement, plusieurs raisons influencent le développement des lymphomes T :
- prédisposition génétique héréditaire;
- influence sur le corps des produits chimiques, des rayons ultraviolets et des rayons pendant une longue période;
- processus inflammatoires dans le corps;
- immunodéficience héréditaire.
Les personnes âgées sont en danger. La combinaison de tous ces facteurs et un stress constant, un surmenage du corps et une mauvaise alimentation peuvent entraîner des modifications des cellules des tissus. Le résultat est la formation de lymphomes à cellules T de la peau ou de néoplasmes cellulaires périphériques.
Symptômes et diagnostic de la maladie
Selon les signes d'évolution, le lymphome T cellulaire se situe à 4 stades :
- La pathologie ne touche qu'une seule zone des ganglions lymphatiques.
- La pathologie ne se produit que d'un côté du diaphragme, dans les nœuds spirituels.
- Dommages bilatéraux au diaphragme.
- Les cellules modifiées se développent et se propagent dans tout le système lymphatique, affectant les organes humains vitaux.
La maladie au quatrième stade peut former des métastases dans le foie, l'estomac, les reins et la moelle osseuse. La maladie apparaît souvent à la suite d’une pathologie avancée aggravée.
Les symptômes de la modification des lymphocytes T peuvent varier :
- degré élevé de transpiration;
- perte de poids soudaine et prolongée ;
- problèmes digestifs;
- faiblesse générale du corps, irritabilité et somnolence;
- changements de température corporelle avec des écarts par rapport à la norme dans un sens ou dans l'autre.
Dans les lymphomes cutanés, des nodules, des taches et des éruptions cutanées de diverses formes apparaissent dans les cellules T. 
Si vous détectez des signes de modifications des leucocytes, vous devez immédiatement consulter un médecin. Au centre d'oncologie, un premier examen par un oncologue est réalisé.
La prochaine étape du diagnostic de la maladie est la morphologie complète de la maladie. Un test d'urine et la détection d'anticorps dans les plasmocytes sont nécessaires.
Un examen complet d'un patient présentant des modifications suspectées des leucocytes T comprend un ordinateur, une imagerie par résonance magnétique et une échographie.
Le pronostic final du lymphome repose sur un examen complet et dépend du type de lésion. Les tumeurs agressives nécessitent un traitement immédiat. Le programme de traitement de ces tumeurs comprend principalement la chimiothérapie et la radiothérapie. Un résultat satisfaisant est une rémission positive après radiothérapie.
Ces informations sont destinées aux professionnels de santé et pharmaceutiques. Les patients ne doivent pas utiliser ces informations comme avis ou recommandations médicales.
Immunodéficiences à cellules T
Les déficits primaires en lymphocytes T sont des troubles héréditaires rares qui affectent le développement et le fonctionnement des lymphocytes T. Ces troubles apparaissent généralement pendant la petite enfance ou la petite enfance ; cependant, l’âge d’apparition des symptômes peut varier en fonction de l’anomalie génétique sous-jacente. Bien que les réponses immunitaires des lymphocytes T puissent être perturbées de manière sélective, des fonctions anormales des lymphocytes B y sont souvent associées, en partie à cause de défauts intrinsèques des lymphocytes B, mais également parce que la majeure partie de la production d’anticorps dépend de l’assistance des lymphocytes T.
SYNDROMES D'IMMUNODÉFICIENCE COMBINÉE SÉVÈRE
Le syndrome d'immunodéficience combinée sévère (SCID) est un trouble héréditaire chez les enfants caractérisé par des fonctions de lymphocytes T et de lymphocytes B profondément défectueuses ou absentes. Le SCID est souvent mortel au cours de la première année de vie, malgré une transplantation thérapeutique de cellules souches ou, en cas de déficit en adénosine désaminase (ADA), un remplacement enzymatique. L’identification précoce des patients affectés avant qu’ils ne développent des infections opportunistes est essentielle pour obtenir une issue favorable. Le diagnostic de SCID est confirmé lorsque l'enfant atteint présente une lymphopénie (Malgré l'hétérogénéité génétique présente, les patients atteints de SCID présentent de nombreuses similitudes au cours des 6 premiers mois de la vie.
Généralement, les enfants affectés peuvent développer les infections suivantes :
Bactérien
Sepsis à Gram négatif
Dissémination du Bacille Calmette-Guérin après vaccination
Causée par des champignons et des protozoaires
Candidose
Aspergillose
Pneumocystis carinii
Viral
Cytomégalovirus
Virus parainfluenza
Adénovirus
Virus respiratoire syncytial
Varicelle disséminée (varicelle)
Polymyélite paralytique acquise par le vaccin
Molluscum contagiosum
Une perte de poids secondaire à une diarrhée ou à une malabsorption peut également survenir. L'apparition d'une éruption érythémateuse maculopapulaire précoce qui ne répond pas au traitement médical peut indiquer une maladie chronique du greffon contre l'hôte (GVHD) résultant d'une transplantation maternelle de lymphocytes T. La plupart des patients atteints de DICS présentent une hypoplasie thymique et des ganglions lymphatiques et du tissu amygdalien absents ou petits et sous-développés ; Une hépatosplénomégalie peut être détectée chez les enfants atteints de GVHD maternelle. Les radiographies thoraciques montrent souvent une ombre thymique absente et un léger aspect pulmonaire, malgré la présence de symptômes respiratoires importants.
La plupart des patients SCID ont un nombre de lymphocytes T CD3+ périphériques de 500 cellules/mm3 ou moins (plage normale de 3 000 à 6 500 cellules/mm3) et un nombre variable de lymphocytes B et de lymphocytes tueurs naturels (NK) en fonction de l'anomalie génétique sous-jacente. Le SCID peut être classé en fonction de la présence ou de l'absence de cellules B et NK en syndromes T-B+NK+, T-B+ NK-, T-B-NK+, T-B-NK- et atypiques T+B+ (Tableau 1) . Les patients présentant un déficit en ADA ont le moins de lymphocytes circulants, tandis que les patients présentant un T+B+ SCID autosomique récessif (AR) inconnu, un déficit en ZAP-70 et une infiltration maternelle de lymphocytes T ont le plus grand nombre de lymphocytes, souvent dans les limites normales. Les patients atteints de SCID réagissent aux tests cutanés d'hypersensibilité de type retardé et les mesures in vitro de la fonction des lymphocytes T sont considérablement réduites à 10 % ou moins des valeurs normales.
En plus du petit nombre de lymphocytes T circulants et d'une hypogammaglobulinémie sévère, on note également une diminution particulière des IgG. Les taux sériques d'IgG dans les limites normales reflètent généralement les anticorps maternels chez les jeunes enfants atteints de SCID ou de gammaglobuline intraveineuse (IVIG). Les taux sériques d'IgA et d'IgM vont de zéro à des valeurs normales liées à l'âge. La présence d'IgE sériques détectables et d'éosinophilie survient généralement chez les enfants atteints de GVHD maternelle ou du syndrome d'Omenn (OS). Malgré la présence d'immunoglobulines sériques, certains patients SCID ne développent pas d'anticorps spécifiques de l'antigène ; par conséquent, l'utilisation de méthodes dépendantes des anticorps telles que les tests immuno-enzymatiques pour dépister l'exposition à des agents infectieux chez les patients atteints de SCID produit des résultats faussement négatifs ou des résultats faussement positifs en raison de l'administration d'IgIV. Au lieu de cela, la détection directe de l'antigène par immunofluorescence ou polymérisation en chaîne devrait être utilisée pour diagnostiquer l'infection chez les patients immunodéprimés.
Le seul traitement du SCID est la reconstitution de cellules souches hématopoïétiques. Le traitement optimal est la transplantation de moelle osseuse (BMT) ou la transplantation de cellules souches périphériques provenant d'un frère ou d'une sœur tissulaire compatible. Malheureusement, pour la plupart des patients, il n’existe pas de donneur familial HLA identique. Une BMT hapto-identique appauvrie en lymphocytes T provenant des parents est souvent réalisée et réussit pour de nombreux patients atteints de SCID. Une greffe de cellules souches de BMT ou de sang de cordon ombilical non apparenté a également été utilisée pour reconstituer le système immunitaire. Les principales complications de la transplantation sont le rejet du greffon, la GVHD, les infections et la toxicité de la chimiothérapie. Certains patients nécessitent une immunosuppression à long terme pour contrôler la GVHD ou des IgIV à vie si la greffe de cellules B du donneur échoue et que la fonction normale des cellules B n'est pas restaurée.
Syndrome d'immunodéficience combinée sévère lié à l'X
Le SCID lié à l'X (XSCID) représente 30 à 40 % de tous les cas de SCID et on pense qu'il survient dans 1 à 2 pour 100 000 naissances. Son mode de transmission a été déduit d'un grand nombre de pedigrees dans lesquels des garçons des générations suivantes sont morts dans la petite enfance à la suite d'infections virales ou fongiques incontrôlables. Le défaut du XSCID a été identifié en 1993 comme une chaîne g commune (gc) du récepteur de l'interleukine (IL)-2. Des études ultérieures ont montré que gc fait également partie des récepteurs de quatre cytokines supplémentaires : IL-4, IL-7, IL-9 et IL-15. La molécule gc semble indispensable à la transmission intracellulaire des signaux d'activation issus des interactions cytokine-récepteur de cytokine, nécessaires à la prolifération et à la maturation des lymphocytes. L'absence de complexes récepteurs contenant des gc se manifeste par une suspension précoce du développement des lymphocytes T et des lymphocytes NK et par la production de lymphocytes B immatures, qui provoquent une commutation isotypique défectueuse nécessaire à la production d'IgG, IgA et IgE. L'interaction entre l'IL-7 et son récepteur est particulièrement critique pour la différenciation des cellules progénitrices lymphoïdes engagées des cellules souches pluripotentes. Ce déficit immunitaire semble être la forme prototypique TB+NK du SCID.
La plupart des patients de sexe masculin atteints de XSCID ont un nombre absolu de lymphocytes inférieur à 2 000 cellules/mm3 dans le sang périphérique, avec moins de 200 cellules/mm3 de lymphocytes T CD3+ (plage de 0 à 800 cellules/mm3), moins de 100 cellules/mm3 de cellules NK. et un pourcentage accru (souvent > 75 %) de lymphocytes B (Tableau 2). Les taux sériques d'IgG et d'IgA sont extrêmement faibles et la production d'anticorps spécifiques est absente. À l’inverse, les taux sériques d’IgM et d’IgE peuvent sembler normaux suite à l’insertion de lymphocytes T maternels. In vitro, les fonctions des lymphocytes T et des lymphocytes NK sont généralement faibles. La plupart des enfants atteints de XSCID ont des lymphocytes T maternels détectés par typage HLA dans leur sang. Une GVHD manifeste peut se développer s’il existe un nombre important de cellules T maternelles capables de réagir aux antigènes HLA acquis par les parents. La présence d'une lymphocytose, de taux normaux d'IgM et d'IgE, d'une hépatomégalie, d'une lymphadénopathie et d'une éruption cutanée chronique résultant d'une GVHD maternelle retarde souvent la détection du XSCID.
Plusieurs mutations différentes du gène IL2RG ont été identifiées chez la progéniture XSCID. Contrairement à la fibrose kystique, aucune mutation courante ne se produit dans le XSCID ; cependant, certains points chauds ont été identifiés dans un gène dans lequel des mutations sont souvent identifiées. Il a été démontré que certaines mutations ont des effets relativement mineurs sur la fonction gc ; ces patients ont tendance à avoir moins de lymphopénie, une fonction des lymphocytes T mieux préservée et des taux sériques d'Ig plus normaux. Ces enfants sont souvent classés comme ayant un AR T+ B+ SCID inconnu, retardant ainsi la reconnaissance de leur anomalie génétique sous-jacente. Étant donné que la plupart des familles XSCID contiennent des mutations individuelles, des analyses séquentielles des régions codantes du gène IL2RG doivent être effectuées pour caractériser les mutations délétères. Le séquençage de l'ADN maternel peut également être effectué pour confirmer les mutations ; cependant, plus de 50 % des patients de sexe masculin atteints de XSCID présentent une mutation IL2RG spontanée sans preuve d'une mutation héréditaire du chromosome X maternel par séquençage de l'ADN ou d'un modèle ouvert d'inactivation du chromosome X. L'HIC histocompatible ou haploidentique ou la greffe de cellules souches de cordon ombilical ou périphérique assurent une reconstitution immunitaire chez la plupart des garçons atteints de XSCID. La VMT in utero traite avec succès les fœtus atteints, et la thérapie génique semble sur le point de devenir une réelle possibilité
Déficit enzymatique JAK3
Les filles et certains garçons présentant le phénotype typique T-B+NK-SCID ne présentent pas de mutations dans le gène IL2RG. Ces patients, dont beaucoup sont nés de parents consanguins, présentent plutôt un AR SCID provoqué par des mutations des deux allèles de l'enzyme JAK3. Le déficit enzymatique JAK3 a été décrit pour la première fois en 1995 et pourrait représenter 10 à 20 % de tous les cas de SCID. JAK3 est une tyrosine kinase cytoplasmique associée à gc et nécessaire à la transduction des signaux liés aux cytokines à partir des récepteurs de cytokines contenant gc. Les mutations de JAK3 interfèrent avec la transmission du signal via ces récepteurs, confortant l'observation selon laquelle la fonction gc dépend absolument de l'activation des JAK3 en aval. Les mutations JAK3 varient et ont tendance à être uniques à chaque famille. Beaucoup excluent l’expression de l’ARNm ou de la protéine JAK3, mais des exceptions ont été décrites, se manifestant par un phénotype SCID plus doux et la production de certaines cellules T et NK.
Déficit en récepteur de l'interleukine-7
Les défauts de la chaîne A du récepteur de l'IL-7 (IL-7Ra) sont des causes rares d'AR SCID. Le phénotype du déficit en IL-7Ra est similaire à celui du déficit en XSCID et JAK3, à quelques exceptions près. Contrairement à ces défauts immunitaires, le déficit en IL-7Ra n’arrête pas le développement des cellules NK et est considéré comme une forme T-B-NK+ de SCID.
Déficit en interleukine-2
Des familles individuelles présentant des défauts dans la production d'IL-2 ont été publiées. Les patients présentant un déficit en IL-2 ont un nombre de lymphocytes périphériques (T+B+ SCID) et une hypogammaalbuminémie relativement normaux. In vitro, la fonction des lymphocytes T est réduite, mais peut être corrigée par l'ajout d'IL-2 recombinante. Les défauts génétiques sous-jacents chez ces patients sont inconnus, mais on pense qu’ils affectent la régulation transcriptionnelle du gène IL-2. Le traitement parentéral par IL-2 recombinante peut permettre une reconstitution immunitaire partielle chez ces patients.
Déficit RAG 1 et 2
Les enfants présentant des anomalies dans les gènes activant la recombinase spécifique des lymphocytes (RAG) 1 ou 2 (RAG1 et RAG2) ont été décrits pour la première fois en 1996. RAG 1 ou RAG 2 se manifeste comme la forme primaire de T-B-NK+ SCID et peut représenter 10 à 20 % de tous les cas. La fonction de RAG 1 et RAG 2 est indispensable pour les générations de récepteurs d’antigènes des lymphocytes T et des lymphocytes B (TCR et BCR, respectivement). Au cours de l'ontogenèse des lymphocytes T et des lymphocytes B, l'ADN chromosomique contenant divers segments variables (V), divergents (D) et accessoires (J) des gènes d'immunoglobuline TCR est réorganisé pour produire des récepteurs d'antigènes fonctionnels. La recombinaison V(D)J confère la diversité des récepteurs antigéniques et la capacité du système immunitaire humain à répondre à plus de 108 antigènes. L'incapacité d'effectuer la recombinaison V(D)J se manifeste par l'arrêt de la maturation des lymphocytes T et des lymphocytes B au stade précoce de la différenciation lymphocytaire avec l'absence totale de tous les lymphocytes T et B et l'agammaglobulinémie. Les cellules NK n'expriment pas de récepteurs spécifiques de l'antigène et leur développement n'est pas altéré par une fonction RAG-1 ou RAG-2 défectueuse. Des mutations dans RAG1 et RAG2 ont été identifiées chez la progéniture d'individus atteints de T-B-NK+ SCID. Des mutations sévères de RAG1 se développent dans des fragments protéiques internes et peuvent être plus fréquentes que des anomalies de RAG2.
Syndrome de Présage (OS)
La SG est un trouble rare de la RA, décrit en 1965 par Omenn sous le nom de SCID, caractérisé par les symptômes suivants :
Données physiques
Érythrodermie
Lymphadénopathie
Hépatosplénomégalie
Incapacité à prendre du poids secondaire à la diarrhée
Œdème généralisé
Fièvre
Données de laboratoire
Hypoalbuminémie
Éosinophilie (> 1 000 cellules par mm3)
Nombre variable de lymphocytes
Diminution à augmentation du nombre de lymphocytes T CD3+
Absence de cellules B
Nombre normal de cellules NK
Fonction des lymphocytes T et des lymphocytes B clairement défectueuse
Hypogammaglobulinémie
Diminution significative des niveaux d'IgG, d'IgM et d'IgA
Hyper-IgE (> 1 000 UI/mL)
En 1998, des mutations de RAG1 ou RAG2 ont été identifiées chez des patients atteints de SG, entraînant une activité partielle de la recombinase V (D) J et le développement de cellules T oligoclonales peu activées, mais anergiques.
Ces manifestations cliniques sont caractéristiques de la SG et la distinguent des autres formes de SCID. Pendant de nombreuses années, on a pensé que la SG était une variante grave de la GVHD maternelle, mais les lymphocytes T maternels intégrés n'ont pas pu être identifiés. Chez les enfants atteints, le nombre de lymphocytes T périphériques variait entre des niveaux significativement réduits et des niveaux normaux. Dans de nombreux cas, le nombre de lymphocytes T CD3+ était normal, mais il n’y avait pas de lymphocytes B circulants. Les immunoglobulines sériques étaient indétectables, à l'exception de taux d'IgE significativement élevés (souvent > 1 000 UI/mL). Comme pour le déficit complet en RAG 1 ou 2, le nombre de cellules NK reste normal chez les patients atteints de SG. L'OS est considérée comme une forme T-B-NK+ de SCID, mais la présence de lymphocytes T oligoclonaux, qui se développent en raison de rares événements de recombinaison V(D)J réussis, brouille le diagnostic. L'hyper IgE est causée par la présence dans les tissus non lymphoïdes de cellules B rares, qui sont stimulées pour produire des IgE par des cellules auxiliaires visant à produire de l'IL-4 et de l'IL-5 et que l'on trouve généralement dans les cas d'hypersensibilité ou de maladies allergiques. La présence d'une lymphadénopathie distingue la SG des autres formes de SCID sans GVHD maternelle. L'examen histologique des ganglions lymphatiques dans la SG révèle des troubles architecturaux, un manque de formation de follicules et une infiltration excessive d'éosinophiles, d'histiocytes et de cellules T activées. La peau est également massivement infiltrée de cellules inflammatoires.
L'analyse du séquençage de l'ADN des patients atteints de SG révèle des mutations dans RAG1 ou RAG2 qui n'éliminent pas complètement l'activité de la recombinase V(D)J. Pour des raisons inconnues, la fonction partielle de RAG1 ou RAG2 entraîne un réarrangement plus productif du TCR que du BCR, de sorte que les lymphocytes T oligoclonaux sont présents et que les lymphocytes B en circulation sont très rares. Il est intéressant de noter que la plupart des enfants touchés sont nés de parents non apparentés. Avant le TDC, la GVHD maternelle doit être exclue. Ces enfants finissent également souvent par tomber gravement malades avec de la fièvre, une entéropathie avec perte de protéines et un œdème généralisé dû à une inflammation des intestins et de la peau. La chimiothérapie ablative et l'immunosuppression sont nécessaires pour prévenir le rejet de greffe par les lymphocytes T autologues activés. La préférence est donnée aux VMT compatibles avec les tissus, car les patients atteints de SG présentent un risque accru d'échec de la transplantation haplo-identique.
Syndrome Navajo combiné sévère
Navajo SCID est une mutation AR survenant chez environ 1 naissance vivante sur 2 000 chez les Amérindiens Ath-Abascan. Le défaut génétique sous-jacent reste inconnu mais est cartographié sur le chromosome 10p lors de l'analyse de la descendance. Les manifestations cliniques du SCID Navajo sont similaires à celles des autres formes de SCID, à l'exception d'un phénomène inhabituel observé chez la plupart des patients au cours des 4 premiers mois de la vie : des ulcères buccaux et génitaux non liés au virus de l'herpès. Les enfants affectés développent une lymphopénie (300 à 1 800 cellules par mm3), avec un nombre de cellules T et B inférieur à 200 cellules par mm3. In vitro, la fonction des lymphocytes T et les taux sériques d'immunoglobulines sont considérablement réduits, tandis que les cellules NK sont abondantes et normales. Navajo SCID est donc classé comme T-B-NK+ SCID, mais n'est pas associé à une fonction RAG-1 ou RAG-2 défectueuse. La présence de l'activité NK complique la VMT, augmentant le risque de rejet de greffe. Un conditionnement ablatif pré-VMT intensif est nécessaire pour réaliser une transplantation chez les enfants affectés.
Déficit en adénosine désaminase
Le déficit en ADA représente environ 15 % de tous les cas de SCID. Il a été décrit pour la première fois en 1972. Contrairement à d’autres formes de SCID, dans lesquelles les mutations affectent spécifiquement les fonctions des lymphocytes T et souvent des lymphocytes B, le déficit en ADA se caractérise par une toxicité métabolique de toutes les cellules, avec les effets les plus prononcés sur les lymphocytes et les progéniteurs lymphoïdes. Le déficit en ADA est la forme la plus courante de T-B-NK-SCID.
L'ADA est une enzyme répandue dans la voie de dégradation des purines qui catalyse la désamination de l'adénosine et de la désoxyadénosine en inosine et désoxyinosine. L'absence d'ADA se manifeste par l'accumulation de ces substrats dans les fluides intracellulaires et extracellulaires. Des taux significativement élevés de désoxyadénosine triphosphate dans les lymphocytes sont associés à l’absorption intracellulaire et à la phosphorylation de la désoxyadénosine, ce qui peut expliquer pourquoi les lymphocytes sont si sensibles aux effets secondaires toxiques de ces métabolites. L'adénosine, la désoxyadénosine et la désoxyadénosine triphosphate inhibent divers processus cellulaires, notamment l'activité de la ribonucléotide réductase, entraînant la mort cellulaire et des lésions tissulaires. L'activité des globules rouges en ADA est généralement mesurée au moment du diagnostic du déficit en ADA, mais elle est généralement inférieure à celle des lymphocytes.
Le spectre clinique et biologique du déficit en ADA est assez large et dépend de la gravité des mutations génétiques sous-jacentes. Le dépistage d'un grand groupe d'adultes en bonne santé a montré que 7 % ou plus de l'activité normale de l'ADA érythrocytaire est associée à une immunité intacte ; Par conséquent, le SCID et les défauts immunitaires moins graves dus au déficit en ADA sont associés à des mutations du gène ADA qui éliminent ou désactivent presque complètement la fonction de l'enzyme. Chez environ 80 % des patients, il présente un déficit classique en ADA d’apparition précoce et se développe au cours des 3 premiers mois de la vie. La caractéristique clinique distinctive chez environ 50 % de ces patients est des anomalies squelettiques, une ventouse initiale et une disparition ou un évasement progressif des jonctions costochondrales visibles sur la radiographie pulmonaire. Ces patients conservent 0,01 % ou moins de l'activité de l'ADA et présentent une alymphocytose (environ 15 à 20 % des cas de déficit en ADA ne sont diagnostiqués qu'à l'âge de 1 à 2 ans ; ces patients sont classés comme ayant un DICS d'apparition tardive retardé causé par des effets moins destructeurs. des mutations du gène ADA. Les enfants atteints conservent 0,1% à 2% d'activité ADA, le nombre de lymphocytes du sang périphérique est inférieur à 500 cellules par mm3, mais une hypoalbuminémie moins sévère au cours de la première année de vie, cependant une diminution du nombre de cellules par mm3. Les lymphocytes T et B et leurs fonctions se produisent assez rapidement et l'infection se développe peu de temps après. L'apparition tardive du déficit en ADA survient chez 5 % ou moins des patients et se caractérise par un diagnostic d'immunodéficience combinée entre 3 et 15 ans, qui est généralement diagnostiqué. précédée d'antécédents d'infection herpétique persistante et d'infections récurrentes des voies respiratoires supérieures, souvent à Streptococcus pneumoniae.
Les maladies auto-immunes, en particulier l'anémie hémolytique et la thrombocytopénie, sont associées à un déficit tardif en ADA. Les résultats de laboratoire sont distinctifs et incluent une activité ADA de 2 % à 5 %, un nombre de lymphocytes circulants inférieur à 800 cellules/mm3, un nombre de lymphocytes T CD3+ inférieur à 500 cellules/mm3 et une éosinophilie. Une hypogammaglobulinémie peut se développer en l’absence d’IgG2 et en cas d’augmentation des taux sériques d’IgE. In vitro, la fonction des lymphocytes T et les réponses anticorps spécifiques, en particulier contre les antigènes polysaccharidiques, sont considérablement réduites. Des familles isolées présentant un début adulte et un déficit partiel en ADA ont été décrites.
Les mesures de l'activité ADA des érythrocytes ne sont pas fiables chez les patients transfusés ; au lieu de cela, la fonction enzymatique dans les lymphocytes ou les fibroblastes doit être mesurée pour confirmer le diagnostic de déficit en ADA. Le diagnostic prénatal du déficit en ADA repose sur le dépistage de l'activité enzymatique dans les cellules obtenues du fœtus, mais doit être accompagné d'un séquençage de l'ADN pour confirmation. Plus de 60 mutations différentes ont été identifiées chez des patients immunodéprimés. La plupart d'entre eux sont regroupés dans des séquences d'ADN codant pour des acides aminés impliqués dans la liaison au substrat ou ayant des fonctions catalytiques. Plus de 50 % des mutations chez les patients présentant un déficit en ADA désactivent complètement l’activité enzymatique et entraînent l’apparition précoce d’un SCID. Contrairement à d’autres formes d’AR SCID, dans lesquelles les patients ont tendance à être homozygotes pour la même mutation, la plupart des patients déficients en ADA sont hétérozygotes mixtes pour deux allèles mutants différents d’ADA.
Le traitement optimal pour le SCID déficient en ADA est le BMT adapté aux tissus. La greffe de BMT haploidentique, même avec une ablation avant la greffe, semble être réduite en cas de déficit en ADA par rapport aux autres diagnostics de SCID. Le taux de mortalité peut également être augmenté. Pour ces raisons, le BMT haplo-identique n’est pas systématiquement réalisé pour traiter le déficit en ADA ; au lieu de cela, les patients affectés en l'absence d'un donneur potentiel compatible avec les tissus sont souvent traités avec une thérapie de remplacement ADA. Le remplacement enzymatique ne nécessite pas l’absorption de l’ADA dans les lymphocytes pour avoir des effets bénéfiques sur les fonctions des lymphocytes T et B. Les métabolites toxiques sont présents dans le liquide extracellulaire et sont en équilibre avec les produits intracellulaires ; par conséquent, la réduction des taux plasmatiques de métabolites due à leur dégradation par l'ADA administrée par voie parentérale entraîne une diminution de la concentration de métabolites intracellulaires. L'ADA de bœuf lié au polyéthylène glucol (PEG) est utilisé depuis 1986 et a permis une reconstitution immunitaire partielle ainsi qu'une amélioration et une survie à long terme chez les patients affectés. Les patients présentant un DICS précoce reçoivent généralement 30 à 60 U/kg/semaine de PEG-ADA ; la dose peut être réduite chez les enfants avec l'âge. Les inconvénients du traitement PEG-ADA comprennent le coût élevé et la nécessité d'une administration fréquente et d'une surveillance étroite des niveaux de métabolites.
La reconstitution immunitaire chez les enfants recevant du PEG-ADA n'est pas complète, avec un nombre et une fonction de lymphocytes B améliorés par rapport à ceux des lymphocytes T. Les patients restent lymphopéniques (le déficit en ADA a été le premier trouble traité par thérapie génique par cellules hématopoïétiques. Certains patients déficients en ADA, dont trois nouveau-nés, ont reçu des lymphocytes T matures autologues corrigés du gène ADA ou des cellules souches de moelle osseuse ou de cordon ombilical. Bien qu'il semble que l'ADA les lymphocytes corrigés présentent des avantages de croissance sélective par rapport aux cellules déficientes en ADA, aucun patient n'a été guéri du SCID ou n'a complètement arrêté de recevoir le traitement par PEG-ADA. De nouvelles études sont prévues et l'efficacité transductive inadéquate des cellules souches hématopoïétiques humaines reste un obstacle important au succès du gène. traitement du déficit en ADA.
IMMUNODÉFICIENCE COMBINÉE SÉVÈRE ATYPIQUE ET IMMUNODÉFICIENCES COMBINÉES
Déficit ZAP -70
Le déficit en ZAP -70 est un AR SCID rare qui a été décrit pour la première fois en 1994 parmi des familles consanguines isolées. La lymphocytose (4 000 à 20 000 cellules par mm 3), l'excès (55 à 75 %) de CD 3+ CD 4+ et moins de 5 % de CD 8+ sont caractéristiques de cette nouvelle forme de T+B+ SCID. La plupart des patients affectés présentent une diminution des taux sériques d'IgG, une altération de la réponse des lymphocytes B et une absence de fonction des lymphocytes T, y compris l'incapacité de l'enfant à rejeter une greffe de peau allogénique.
L'absence de lymphocytes T CD 8+ est associée à des mutations de la protéine tyrosine kinase, ZAP -70. La fonction ZAP -70 est essentielle à la transmission du signal du TCR et son absence entraîne une différenciation anormale des thymocytes et une activation défectueuse des lymphocytes T nécessaires à la prolifération et au fonctionnement des lymphocytes T. À ce jour, moins de 15 patients déficients en ZAP ont été décrits, la plupart d'entre eux présentent des mutations dans une faible proportion. ZAP-70 gènes qui affectent la stabilité des protéines et la fonction catalytique. Le séquençage de l'ADN est nécessaire pour confirmer l'héritage des deux mutants ZAP-70 allèles chez les patients SCID caractérisés par un manque de cellules CD 8 + T circulantes.
Lacunes CD 3
Les TCR sont assemblés et exprimés en surface en association avec le CD 3, qui est un complexe de six sous-unités (par exemple, ed et xx). Deux formes de déficit en CD 3 ont été décrites dans des familles distinctes dans lesquelles elles sont provoquées par des mutations de la protéine CD 3, entraînant une expression défectueuse du TCR. Bien qu'elles ne soient pas aussi graves que les défauts de recombinaison, les mutations de CD 3g et CD 3e provoquent des déficits immunitaires légers à modérés. Les patients affectés ont un nombre et une fonction réduits de lymphocytes T ; Les cellules B sont affectées à des degrés divers.
Syndromes lymphocytaires purs
Les syndromes lymphocytaires purs (SBL) ressemblent aux déficits immunitaires sélectifs des lymphocytes T, mais chez certains patients, ils ne peuvent pas être distingués de ceux du SCID. Les deux types de BLS reflètent une expression déficiente de molécules de classe du complexe majeur d'histocompatibilité (CMH) (HLA A, BC) ou de classe II (HLA DR, DQ ou DP) sur les cellules hématopoïétiques. Un diagnostic de déficit en BLS de type 1 ou en CMH de classe I est suggéré lorsque les molécules HLA A, B et C présentes sur les lymphocytes ne peuvent pas être détectées par les méthodes sérologiques. Ce trouble a été identifié chez un petit nombre de familles consanguines et sa présentation clinique est variable. Contrairement au SBL de type 2, la plupart des patients atteints du SBL de type 1 sont asymptomatiques pendant l'enfance ; bien que des infections respiratoires persistantes et des maladies pulmonaires chroniques se développent chez ces enfants au cours des dix premières années de leur vie. Le diagnostic est retardé car les patients ont un nombre normal de cellules T et B périphériques et des fonctions immunitaires relativement préservées. Le phénotype lymphocytaire peut révéler une diminution des cellules CD 8+T.
Le BLS de type 1 est associé à des mutations dans l’un des nombreux gènes spécifiques. Certains patients présentent des défauts dans la transcription des gènes du CMH de classe 1, tandis que d'autres présentent des mutations dans les gènes codant pour le traitement de l'antigène du transporteur associé (TAP)-1 ou TAP-2. Ces protéines sont impliquées dans le transport des antigènes transformés du cytoplasme vers le réticulum endoplasmique. En l'absence de TAP-1 ou de TAP-2, les molécules du CMH de classe I dans le réticulum endoplasmique sont incapables de se charger en antigènes, ce qui se manifeste par leur dégradation et une diminution de leur expression à la surface cellulaire. Le traitement du BLS de type 1 est favorable et le VMT n’est généralement pas indiqué.
Le déficit en BLS de type 2 ou CMH de classe II est une forme assez rare d'AR T+B+ SCID qui touche principalement les enfants nés dans des familles consanguines d'origine maghrébine ou méditerranéenne. Les patients présentant un déficit en CMH de classe II développent souvent une maladie hépatique associée à une infection chronique à Cryptosporoium. Les patients ont un nombre normal de lymphocytes circulants avec un nombre réduit de lymphocytes T CD 4 +, une hypogammaglobulinémie et une prolifération in vitro significativement défectueuse des lymphocytes T et des lymphocytes B en antigènes spécifiques.
Une caractéristique du BLS de type 2 est la capacité réduite des lymphocytes à stimuler les lymphocytes allogéniques dans les cultures in vitro. Cette observation est étayée par la découverte selon laquelle les molécules du CMH de classe II sont exprimées à moins de 5 % de l'intensité normale sur les cellules hématopoïétiques des patients affectés ; cependant, l'analyse de séquençage de l'ADN n'a pas révélé de mutations dans les gènes du CMH de classe II ; les patients ont plutôt hérité de mutations dans l'un des nombreux gènes importants dans leur transcription, notamment CIITA, RFX-5, RFX-B ou RFX-AP. Le pronostic à long terme pour les patients atteints de BLS de type 2 est désastreux. La plupart des patients meurent d’une défaillance progressive d’un organe. En règle générale, les VMT compatibles avec les tissus ou haplo-identiques ne permettent pas une reconstitution immunitaire ni une prolongation de la survie pendant la transition vers l'âge adulte.
Déficit en purine nucléoside phosphorylase
Le déficit en purine nucléoside phosphorylase (PNP) est un déficit immunitaire combiné rare associé à des défauts immunitaires et à des symptômes neurologiques, notamment des retards de développement importants, des problèmes de comportement et des anomalies motrices. Le SCID présentant des déficits neurologiques doit être considéré comme présentant un déficit en PNP jusqu'à preuve du contraire. Les manifestations cliniques du déficit en PNP sont essentiellement les mêmes que celles du SCID ; cependant, la PNP est souvent classée comme un trouble d'immunodéficience combinée car les anomalies des lymphocytes B sont relativement bénignes dans la petite enfance. En règle générale, les patients souffrent de lymphopénie (fonction des lymphocytes T in vitro). Le déclin du nombre et de la fonction des lymphocytes B se produit avec le temps, se manifestant par de faibles taux sériques d'IgG et d'IgA. Malgré une diminution significative de la fonction des lymphocytes B, dans plus de 30 % Des patients développent des maladies auto-immunes, telles qu'une anémie hémolytique, un purpura plaquettaire et une vascularite. Certains patients meurent d'un lymphome et d'autres tumeurs.
Le PNP accompagne l'ADA et précède l'hypoxanthine phosphoribosyltransférase dans la voie de dégradation des purines. Il catalyse la phosphorylation de l'inosine-désoxyinosine et de la guanosine-désoxyguanosine en hypoxanthine et en guanine, respectivement. La guanosine et la désoxyguanosine semblent être toxiques pour les lymphocytes T, et une diminution des taux de guanosine triphosphate (GTP) intracellulaire peut être principalement dommageable pour le système nerveux central. Le manque de fonction PNP est généralement démontré par un taux d’acide urique sérique inférieur à 1 mg/dL, qui peut être utilisé pour dépister ce trouble. La mesure de l'activité PNP érythrocytaire permet de confirmer le diagnostic. Le séquençage de l'ADN a révélé l'héritabilité des mutations individuelles dans la plupart des familles. De nombreux patients présentant un déficit en PNP sont nés de parents consanguins et présentent des mutations qui éliminent complètement l’expression des protéines et l’activité enzymatique. Les perspectives concernant le déficit du PNP sont extrêmement mauvaises. Seul un petit nombre de patients ont réussi une greffe de cellules souches hématopoïétiques pour le BMT. La GVHD est une complication importante et il n'y a aucune amélioration des symptômes neurologiques après la VMT. Le remplacement enzymatique et la thérapie génique du déficit en PNP ne sont pas actuellement disponibles.
Ataxie télangiectasie et variantes
L'ataxie télangiectasie (AT) est un trouble AR complexe caractérisé par une ataxie cérébrale, une télangiectasie oculocutanée, une radiosensibilité cellulaire, une susceptibilité à la malignité et un déficit immunitaire combiné. La prévalence mondiale de l'AT est estimée à 3 naissances vivantes sur 10 6. Le produit génique muté AT (ATM) a été identifié en 1995 et est une protéine kinase de signalisation impliquée dans le contrôle du cycle cellulaire, la recombinaison de l'ADN, l'apoptose et d'autres réponses cellulaires aux dommages de l'ADN.
Le principal symptôme de l'AT est une déficience neurologique, se manifestant généralement par une démarche ataxique au cours de la deuxième année de vie, mais observée chez plus de 84 % des patients à l'âge de 4 ans. L'ataxie affecte progressivement le tronc, les membres et les muscles palatins, se manifestant par une dysarthrie d'élocution, une bave, une apraxie oculaire, une choréoathétose et une incapacité de bouger librement dès la 10e année de vie. Les télangiectasies de la sclère et de la peau se développent ensuite entre 4 et 8 ans. L'immunité cellulaire et humorale dans l'AT varie, mais conduit souvent à des infections récurrentes des voies respiratoires supérieures et inférieures et à des maladies pulmonaires chroniques. Une incidence accrue d'infections bactériennes et virales est observée chez la plupart des patients AT âgés de 3 à 6 ans et diminue légèrement avec le traitement par IgIV. Une caractéristique importante de l'AT est une prédisposition prononcée aux néoplasmes à cellules T et B, en particulier à la leucémie et aux lymphomes hodgkiniens et non hodgkiniens. Les tumeurs malignes constituent la deuxième cause de décès chez les patients atteints d'AT, devant les maladies pulmonaires et la pneumonie d'aspiration.
L'instabilité génétique, se manifestant par des translocations chromosomiques et des cassures excessives de l'ADN, est une caractéristique cardinale de l'AT et peut être une cause de cancer et d'immunodéficience chez les enfants affectés. Les manifestations cliniques supplémentaires de l'AT sont un retard de croissance, un hypogonadisme, un début tardif de la puberté et un diabète sucré non cétonique insulino-résistant. Des taux sériques élevés d'a-fœtoprotéine (AFP) et d'antigène carcinoembryonnaire sont assez courants et peuvent être utiles pour établir le diagnostic.
Les patients atteints d'AT présentent une lymphopénie progressive affectant à la fois les lymphocytes T et les lymphocytes B, y compris la perte sélective des lymphocytes T CD 4 + avec une inversion du rapport CD 4 - CD 8 normal. Un déclin de la fonction des lymphocytes T et des lymphocytes B se développe avec le temps et on pense. être associé à une formation excessive de cassures de l'ADN au sein du gène TCR et de l'immunoglobuline sur les chromosomes 7 et 14, respectivement. Les patients AT présentent des réponses prolifératives in vitro réduites des lymphocytes T, mais la gravité du dysfonctionnement immunitaire varie. Les défauts humoraux comprennent des déficits en IgA et IgE chez la plupart des patients AT et une diminution des taux d'isohémagglutinines et d'IgG 2 sériques chez une minorité d'individus. Les réponses en anticorps spécifiques peuvent également être altérées.
Une sensibilité accrue aux rayonnements ionisants et une instabilité génétique sont assez courantes chez les patients atteints d'AT. L'ATM peut agir en activant des repères du cycle cellulaire en réponse à des dommages à l'ADN, y compris des événements de recombinaison V )D )J normaux dans les cellules T et B. Les anomalies dans le contrôle du cycle cellulaire médié par l'ATM n'empêchent pas la synthèse de l'ADN de se poursuivre malgré les dommages à l'ADN, comme en témoigne l'accumulation de débris chromosomiques au fil du temps et la mort cellulaire. Les thymocytes, les lymphocytes B immatures et les cellules du système nerveux central et de l'endothélium vasculaire pourraient être plus sensibles à ces phénomènes.
La plupart des patients AT sont hétérozygotes pour deux mutations ATM différentes affectant la stabilité des protéines. Malheureusement, la radiosensibilité cellulaire ne permet pas la transplantation comme option de traitement viable pour l'AT. De plus, les protocoles de chimiothérapie standards ne peuvent pas être utilisés pour traiter les tumeurs malignes associées à l'AT. Certains patients AT survivent plusieurs années avec des tumeurs lymphoïdes, mais le pronostic à long terme est désastreux. La plupart des patients atteints d’AT ne survivent pas au-delà de la troisième décennie de leur vie. Les individus hétérozygotes AT peuvent également être prédisposés aux tumeurs.
Les variantes AT incluent le syndrome de Nimègue (NBS), qui est également caractérisé par des défauts immunitaires des lymphocytes T et B, une radiosensibilité, une instabilité génétique et une susceptibilité au cancer. Le NBS diffère de l'AT par la présence d'une microcéphalie chez plus de 75 % des enfants atteints, d'un léger retard mental et d'une dysmorphie faciale, ainsi que de l'absence de télangiectasie, d'ataxie progressive et de taux élevés d'AFP. Le gène muté chez les patients NBS code pour la nibrine, une protéine de type ATM impliquée dans la réparation de l'ADN et le contrôle du cycle cellulaire. L'immunodéficience chez les patients atteints de NBS est assez importante et comprend une lymphopénie, une diminution du nombre de cellules CD 4 + et une inversion du rapport CD 4-CD 8, une augmentation du nombre de cellules NK et une diminution de la fonction des cellules T in vitro. Des IgG et IgA sériques faibles ou absentes avec des taux d'IgM normaux ou élevés surviennent chez plus de 90 % des patients. Un déficit en IgG 2 et une production défectueuse d'anticorps anti-pneumococciques peuvent également se développer.
Syndrome de Wiskott-Aldrich
Le syndrome de Wiskott-Aldrich (WAS) est un trouble d'immunodéficience héréditaire lié à l'X caractérisé par de l'eczéma, une thrombocytopénie congénitale avec petites plaquettes et des infections récurrentes. Elle touche environ 1 à 2 enfants sur 10 6. Le produit du gène WAS a été identifié comme étant la protéine WAS (WASP) en 1994. Il a également été démontré par la suite que la thrombocytopénie héréditaire liée à l'X (XLT) résultait de mutations dans WASP. WASP est une molécule de signalisation intracellulaire qui, grâce à son association avec d'autres protéines, est impliquée dans la conduction du signal TCR, jouant un rôle important dans la régulation de l'action de l'organisation cytosquelettique.
Il existe une forte corrélation entre le génotype et le phénotype dans WAS. Bien que tous les patients présentent des anomalies plaquettaires, les hommes présentant les mutations WAS les plus graves (représentant environ 30 % de tous les cas) courent le plus grand risque de décès par hémorragie, infection, auto-immunité ou tumeur maligne. Les patients atteints de XLT ou de WAS léger peuvent souffrir d'eczéma et de divers déficits immunitaires, mais ne développent pas de cancer ni de maladies auto-immunes. Les patients atteints de WAS sévère ou classique présentent souvent au cours de la période néonatale ou de la petite enfance des pétéchies et des saignements provoqués par une thrombocytopénie (généralement 10 000 à 50 000 cellules par mm3). Une diarrhée sanglante, une hémorragie intracrânienne et un saignement excessif provenant des restes de cordon ombilical ou après la circoncision sont parfois les premières manifestations du WAS classique. Le diagnostic peut être confirmé en déterminant la taille des plaquettes. Contrairement au purpura thrombocytopénique idiopathique, les volumes plaquettaires dans le WAS ont un volume moyen de 3,8 à 5,0 fL (plage normale, 7,1 à 10,5 fL). Bien que WAS ne réponde pas aux stéroïdes ou aux IgIV, une splénectomie peut être réalisée en cas de thrombocytopénie sévère mais augmente le risque de décès lié à la septicémie ; cependant, des crises similaires à celles du purpura thrombocytopénique idiopathique aigu surviennent par la suite, pouvant s'accompagner d'hémorragies potentiellement mortelles. Environ 25 % des décès non liés au BMT dans le WAS sont causés par des complications hémorragiques.
L'eczéma se développe chez plus de 80 % des patients et survient souvent avant 6 mois de vie. Des infections sinopulmonaires récurrentes à micro-organismes encapsulés se développent chez les garçons atteints de WAS classique au cours des 2 premières années de la vie. Des infections moins graves peuvent être observées chez les patients atteints de XLT ou de WAS faible. À mesure que la fonction des lymphocytes T diminue, des infections opportunistes telles que la pneumonie causée par Pneumocystis carinii et les infections récurrentes causées par le virus de l'herpès. Environ 40 à 50 % des décès non liés au BMT dans la région WAS sont causés par des infections.
Chez les patients atteints de WAS classique qui n'ont pas subi de transplantation, la prévalence de la leucémie, des lymphomes du système nerveux abdominal et central et des tumeurs associées au virus Ebstein-Barr (EBV) est significativement augmentée, représentant 25 % de tous les décès non liés au BMT. Les maladies auto-immunes, notamment l'anémie hémolytique, l'arthrite, la vascularite, les maladies inflammatoires de l'intestin et la glomérulonéphrite, sont observées chez environ 40 % des patients naïfs de greffe présentant des mutations graves de WASP et peuvent augmenter le risque de tumeurs malignes ultérieures. Les tumeurs malignes et l'auto-immunité ne surviennent pas chez les patients atteints de XLT ou de WAS faible.
Chez les patients atteints, un nombre normal de lymphocytes du sang périphérique est détecté pendant l'enfance. La lymphopénie (cellules T CD3 + et CD 8 +) est généralement observée à l'âge de 6 ans. À l'inverse, le nombre de cellules B et de cellules NK reste normal. La plupart des patients développent une altération de la fonction des cellules T. Des taux sériques d'IgG normaux et une diminution des IgM en association avec une production défectueuse d'anticorps antipneumococciques et une absence d'isohémagglutinines sont systématiquement constatées chez les patients WAS. Étant donné que les patients WAS présentent des fonctions altérées des lymphocytes T et B, la plupart d'entre eux reçoivent des IVIG.
La greffe de cellules souches hématopoïétiques est la seule option thérapeutique pour le WAS classique. Le plus grand succès de la reconstitution immunitaire et plaquettaire est obtenu chez les enfants qui ont reçu une BMT compatible avec les tissus avant l'âge de 5 ans. La transplantation de cellules souches de sang de cordon ombilical et la BMT non apparentée se sont également révélées efficaces chez les jeunes enfants. L'ablation pré-transplantation est nécessaire pour garantir l'incorporation des cellules T et des plaquettes du donneur. Le rejet de greffe dans le BMT haplo-identique est alarmant, mais les patients WAS qui subissent une transplantation réussie sont guéris de l'immunodéficience et d'autres complications de la maladie. Plus de 100 mutations du gène WASP ont été décrites et bon nombre d’entre elles sont uniques aux familles affectées. Cependant, certains points chauds du gène WAS ont été identifiés, là où les mutations se produisent avec la fréquence la plus élevée. Le WAS classique est généralement causé par des mutations qui suppriment ou éliminent l’expression des protéines.
Trouble lymphoprolifératif lié à l'X
La maladie lymphoproliférative liée à l'X (XLP) ou syndrome de Duncan est un déficit immunitaire combiné qui survient après une exposition aux virus Ebstein-Barr (EBV). La fréquence du XLP est d'environ 1 à 3 garçons sur 10 6. Il a été démontré en 1998 que le gène XLP (LYP) code pour la protéine SAP spécifique des lymphocytes T (protéine associée à la molécule d'activation des lymphocytes de signalisation). SAP joue un rôle important dans la régulation de la transduction du signal dans les cellules T activées, et les cellules T déficientes en SAP ne peuvent pas contrôler la prolifération des cellules B induite par l'EBV ; par conséquent, la capacité réduite du système immunitaire à éliminer les lymphocytes B infectés par l’EBV dans le XLP est associée à des défauts dans les réponses des lymphocytes T spécifiques à l’EBV plutôt qu’à ceux des lymphocytes B.
Avant l’infection par l’EBV, la plupart des garçons porteurs de mutations XLP étaient cliniquement sains. L'exposition à l'EBV entraîne une mononucléose infectieuse fulminante et souvent mortelle dans 60 % des cas. Une hépatite sévère, un syndrome hématophagocytaire associé au virus, une anémie aplasique et une défaillance multiviscérale accompagnée de la mort se développent dans les 8 semaines suivant le début de la mononucléose infectieuse dans 95 % des cas. Des lymphomes hodgkiniens et non hodgkiniens de l'abdomen et du système nerveux central se développent chez environ 30 % des garçons atteints de XLP. Très peu de patients atteints d’une maladie lymphoproliférative associée à l’EBV survivent plus de 10 ans. Les patients atteints de dysgammaglobulinémie, caractérisée par une hypogammaglobulinémie avec des taux sériques élevés d'IgM, ont un pronostic favorable à moins qu'ils ne soient associés à une maladie lymphoproliférative. Le pronostic des patients atteints de XLP est désastreux, la plupart des enfants mourant pendant l'enfance ou l'adolescence. Les médicaments antiviraux ou immunosuppresseurs ne sont pas efficaces dans le traitement des maladies fulminantes. Seul un petit nombre de patients ont connu une reconstitution immunitaire après la transplantation ; cependant, la transplantation est liée à l'âge du patient : la plupart des patients de plus de 15 ans ne survivent pas à la BMT.
Après une infection par l’EBV, les patients XLP présentent des anomalies des lymphocytes T et des lymphocytes B. Bien que les patients aient un nombre normal de lymphocytes T, les lymphocytes T CD 8 + prédominent, avec une inversion du rapport CD 4 - CD 8 normal. In vitro, la fonction des lymphocytes T est variable mais est souvent supprimée par rapport aux réponses normales. Les titres d’anticorps contre l’antigène nucléaire associé à l’EBV sont absents ou significativement réduits dans de nombreux cas, tandis que la production d’anticorps contre l’antigène de la capside varie. Des anomalies de la fonction NK étaient évidentes chez 30 % des garçons affectés.
DÉFAUTS SÉLECTIFS DES CELLULES T
syndrome de DiGeorge
Il a été démontré que plus de 90 % des enfants présentant des malformations cardiaques et cranio-faciales congénitales complexes, qui ont été initialement diagnostiquées comme distinguant divers syndromes génétiques, notamment les syndromes de DiGeorge (DGS), les anomalies faciales vélocardiaques et conotroncales de Shprintzen, présentent une microdélétion commune d'une copie. du chromosome 22. On pense que la microdélétion chromosomique hémizygote 22q 11.2 est associée à de nombreux problèmes médicaux rencontrés par les enfants affectés, notamment un retard de développement et une immunodéficience. Par analyse d'hybridation in situ par fluorescence (FISH), cette anomalie chromosomique peut être diagnostiquée chez n'importe quel enfant et chez les membres de sa famille jusqu'alors non reconnus.
Le DGS implique classiquement des malformations cardiaques conotroncales associées à une hypocalcémie persistante et à un déficit immunitaire cellulaire secondaire à un défaut de développement plus envahissant des troisième et quatrième champs de la poche pharyngée qui affecte les glandes parathyroïdes et le thymus. Les anomalies cardiaques spécifiques les plus souvent associées au DGS sont : l'interruption de la crosse aortique, la tétralgie de Fallot et le tronc artériel. L'expression phénotypique du DGS et d'autres troubles de délétion du chromosome 22q 11.2 varie même au sein des familles et comprend souvent une fente palatine et des anomalies faciales.
Les microdélétions du chromosome 22q 11.2 détectées par FISH varient en taille, mais incluent généralement une séquence liée à la région critique dite de DiGeorge ou au locus DGS -1. Dans environ 25 % des cas, la délétion est héréditaire, souvent acquise d'un parent phénotypiquement sain. Les délétions restantes du chromosome 22q 11.2 surviennent de novo chez les enfants affectés. La prévalence de cette anomalie chromosomique est d'au moins 13 pour 100 000 naissances vivantes, ce qui en fait la cause génétique la plus courante de cardiopathie congénitale ; cependant, 20 % des enfants atteints du syndrome de délétion 22q 11.2 ne présentent pas d'anomalies cardiaques. Une analyse cartographique détaillée nous a permis d'identifier une région critique minimale de DiGeorge, mais le ou les gènes spécifiques responsables du syndrome de délétion du chromosome 22q 11.2 n'ont pas été trouvés. Malgré cela, on considère que ces troubles résultent d’une hoplo-insuffisance de gènes impliqués dans la migration croisée neuronale. Il est certain que, puisque les phénotypes présentés par les enfants atteints du syndrome de délétion 22q 11.2 et les membres de leur famille affectés comprennent un large éventail de signes physiques, de multiples gènes et facteurs modificateurs déterminent leur cause. De plus, de rares patients présentant des manifestations phénotypiques de DGS ne présentent pas de microdélétion détectable du chromosome 22q 11.2, certains de ces patients sont homozygotes pour une délétion du chromosome 10p, qui définit le locus ou locus DGS-II ;
Le spectre des défauts immunitaires dans le DGS est assez large, mais comprend le plus souvent une diminution du nombre de lymphocytes T CD3+, un nombre de lymphocytes T CD4+ inférieur à 1 000 cellules par mm 3 et une immunité cellulaire légèrement altérée. 50 % ou moins du nombre normal de lymphocytes T et 20 % ont des réponses prolifératives de lymphocytes T in vitro inférieures à 50 % de la normale. Certains immunologistes estiment que le diagnostic de DGS devrait être limité aux enfants atteints du syndrome de délétion chromosomique 22q 11.2 qui en ont moins. plus de 500 cellules. Cellules T CD 3+ par mm 3 Les chercheurs pensent généralement que la maturation des lymphocytes T est normale chez les patients atteints de DGS et que la lymphopénie est directement liée à une diminution quantitative de la masse thymique fonctionnelle. résultat immunologique. Le déficit immunitaire s’améliore généralement avec l’âge et de nombreux enfants produisent un nombre normal de lymphocytes T fonctionnels à la fin de la deuxième année de vie ; cependant, le taux et l'ampleur de la récupération immunitaire ne sont pas prévisibles et environ 5 % des patients atteints de DGS ont un nombre et une fonction de lymphocytes T considérablement réduits en raison d'une aplasie thymique ; ces patients peuvent avoir besoin d'une BMT. Cinquante pour cent des patients ont également une immunité humorale altérée. Les conséquences de l'immunodéficience chez les enfants atteints de DGS comprennent une susceptibilité accrue aux infections virales. Cette découverte se traduit en pratique par le report des vaccinations avec des virus vivants jusqu'à ce que la preuve de l'immunocompétence soit disponible. Le critère habituel comprend un nombre normal de lymphocytes T, 75 % ou plus des réponses prolifératives normales des lymphocytes T in vitro et une production d'anticorps spécifiques après l'immunisation contre l'anatoxine tétanique. La présence et la gravité des défauts immunitaires chez les enfants atteints du syndrome de délétion du chromosome 22q 11.2 varient et ne sont pas en corrélation avec d'autres manifestations phénotypiques. De plus, l’immunodéficience des lymphocytes T se développe aussi souvent chez les enfants diagnostiqués avec un syndrome de délétion chromosomique 22q 11.2 non-DGS que chez les enfants présentant des manifestations classiques du DGS.
Jusqu'à récemment, de nombreux enfants atteints du syndrome de délétion chromosomique 22q 11.2 mouraient des suites de leurs malformations cardiaques. Ces enfants survivent désormais jusqu’à l’âge adulte et de nouveaux aspects de leurs maladies chroniques apparaissent. Les manifestations reconnues du syndrome de délétion chromosomique 22q 11.2 sont actuellement considérées comme l'auto-immunité, des difficultés d'apprentissage importantes, un dysfonctionnement comportemental et émotionnel et des diagnostics psychiatriques. Il est important de réaliser une consultation familiale détaillant ces manifestations du syndrome de délétion chromosomique 22q 11.2.
Candidose cutanéo-muqueuse chronique
La candidose cutanéo-muqueuse chronique (CMC) est un groupe hétérozygote de troubles caractérisés par une infection fongique persistante des ongles, de la peau et des muqueuses. Plus de 50 % des patients souffrent d’une maladie AR caractérisée par une polyendocrinopathie et présentent des mutations du gène AIRE codant pour une protéine régulatrice de la transcription. L’anomalie génétique sous-jacente chez les enfants atteints de SMS isolé n’est pas connue. La plupart des patients ont un nombre et une fonction lymphocytaires normaux ; cependant, une constatation distinctive chez certains patients présentant un SMS isolé est l'absence de prolifération in vitro des lymphocytes T en antigène de Candida.
RÉSUMÉ
Les défauts immunitaires des lymphocytes T constituent la majorité des déficits immunitaires héréditaires diagnostiqués pendant l’enfance. La plupart des déficits immunitaires cellulaires sont associés à des défauts humoraux dans diverses manifestations cliniques et biologiques. Les anomalies génétiques sous-jacentes sont connues et définies pour la plupart des anomalies immunitaires héréditaires des lymphocytes T, et l'analyse des mutations fait de plus en plus partie intégrante de l'évaluation et du diagnostic. Une discussion détaillée avec la famille sur la corrélation phénotype-génotype est essentielle à la prise en charge médicale et au pronostic à long terme.
Source : Melisa E. Elder, / IMMUNODÉFICIENCES DES CELLULES T / CLINIQUES PÉDIATRIQUES D'AMÉRIQUE DU NORD VOL.47. N°6.