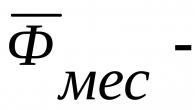Un ensemble de cellules productrices d’hormones uniques est appelé système endocrinien diffus (DES). Parmi les cellules productrices d'hormones uniques, on distingue deux groupes indépendants : I - les cellules neuroendocrines de la série APUD (origine nerveuse) ; II - cellules d'origine non nerveuse.
Le premier groupe comprend les neurocytes sécrétoires formés à partir des neuroblastes de la crête neurale, qui ont la capacité de produire simultanément des neuroamines et également de synthétiser des hormones protéiques, c'est-à-dire ayant les caractéristiques des cellules nerveuses et endocriniennes, donc appelées neuroendocriniencellules. Ces cellules se caractérisent par leur capacité à capter et à décarboxyler les précurseurs aminés.
Selon les concepts modernes, les cellules APUD se développent à partir de toutes les couches germinales et sont présentes dans tous les types de tissus :
1) dérivés du neuroectoderme (cellules neuroendocrines noyaux neurosécréteurs de l'hypothalamus, de la glande pinéale, de la médullosurrénale, des neurones leptidergiques du système nerveux central et périphérique) ; 2) dérivés de l'ectoderme cutané (cellules de la série APUD de l'adénohypophyse, cellules de Merkel de l'épiderme) ; 3) dérivés de l'endoderme intestinal - entérinocytes - cellules du système gastro-entéropancréatique ; 4) dérivés du mésoderme (les cardiomyocytes sécréteurs se développent à partir de la plaque myoépicardique) ; 5) dérivés du mésenchyme - mastocytes
Les cellules de la série APUD se caractérisent par les caractéristiques suivantes : la présence de granules spécifiques, la présence d'amines (catécholamines ou sérotonine), l'absorption d'acides aminés - précurseurs d'amines, la présence d'une enzyme - décarboxylase de ces acides aminés.
Les cellules de la série APUD se trouvent dans le cerveau et dans de nombreux organes – endocriniens et non endocriniens. Les cellules de la série APUD se trouvent dans la plupart des organes et systèmes - dans le tractus gastro-intestinal, le système génito-urinaire, la peau, les organes endocriniens (glande thyroïde), l'utérus, le thymus, les paraganglions, etc.
Sur la base de caractéristiques morphologiques, biochimiques et fonctionnelles, plus de 20 types de cellules APU de la série D ont été identifiés, désignés par les lettres de l'alphabet latin A. B, C, D, etc. Il est d'usage de classer les cellules endocriniennes de la système gastro-entéropancréatique dans un groupe spécial.
Une description des cellules endocriniennes de divers organes est donnée dans les chapitres correspondants.
Des exemples de cellules neuroendocrines de ce groupe situées dans les organes endocriniens sont les cellules parafolliculaires de la glande thyroïde et les cellules chromaffines de la médullosurrénale, et dans les cellules non eidocrines - l'entéronite (cellules entérochromaffines) dans la membrane muqueuse du tractus gastro-intestinal.
Les hormones odigopelt produites par les cellules neuroendocrines ont un effet local sur les cellules des organes dans lesquels elles sont localisées. mais principalement distant (endocrinien) - sur les fonctions générales du corps jusqu'à l'activité nerveuse supérieure
Une caractéristique topographique commune de ces cellules est leur emplacement à proximité des vaisseaux sanguins.
Le rapport de formation d'oligopeptides régulateurs et de neuroamines dans différentes cellules neuroendocrines peut être différent.
Les cellules endocriniennes de la série APUD montrent une dépendance étroite et directe à l'influx nerveux qui leur parvient par innervation sympathique et parasympathique, mais ne répondent pas aux hormones de l'hypophyse antérieure ; leur état et leur activité après hypophysectomie ne sont pas affectés.
Le deuxième groupe comprend les cellules productrices d’hormones uniques ou leurs amas, provenant de sources autres que les neuroblastes. Ce groupe comprend une variété de cellules d'organes endocriniens et non endocriniens qui sécrètent des stéroïdes et d'autres hormones : l'insuline (cellules B), le glucagon (cellules A), l'entéroglucine (cellules L), les peptides (cellules D, K- cellules), la sécrétine (cellules S), etc. Il s'agit également des cellules de Leydig (glandulocytes) des testicules, produisant de la testostérone, et des cellules de la couche granuleuse des follicules ovariens, produisant des œstrogènes et de la progestérone, qui sont des hormones stéroïdes (ces cellules sont d'origine mésodermique). La production de ces hormones est activée par les gonadotrophines adénopituitaires et non par l'influx nerveux.
En 1968, le pathologiste et histochimiste anglais E. Pierce a étayé la théorie de l'existence dans le corps d'un système cellulaire neuroendocrinien spécialisé et hautement organisé, dont la principale propriété spécifique est la capacité de ses cellules constitutives à produire des amines biogènes et des hormones polypeptidiques. (Système APUD). Les cellules incluses dans le système APUD sont appelées apudocytes. Le nom du système est une abréviation de mots anglais (amin - amines ; précurseur - prédécesseur ; absorption - accumulation ; décarboxylation - décarboxylation), indiquant l'une des principales propriétés des apudocytes : la capacité à former des amines biogènes par décarboxylation de leurs précurseurs accumulés. . En fonction de la nature de leurs fonctions, les substances biologiquement actives du système sont divisées en deux groupes : 1) les composés qui remplissent des fonctions spécifiques strictement définies (insuline, glucagon, ACTH, hormone de croissance, mélatonine, etc.) et 2) les composés avec fonctions diverses (sérotonine, catécholamines, etc.) . Ces substances sont produites dans presque tous les organes. Les apudocytes agissent au niveau tissulaire en tant que régulateurs de l'homéostasie et contrôlent les processus métaboliques. Par conséquent, avec la pathologie (apparition d'apuds dans certains organes), se développent les symptômes d'une maladie endocrinienne, correspondant au profil des hormones sécrétées.
L'activité du système APUD, localisée dans les tissus des poumons et du tractus gastro-intestinal (estomac, intestins et pancréas), est aujourd'hui la plus étudiée.
Les apudocytes dans les poumons sont représentés par les cellules de Feyter et de Kulchitsky. Ils sont plus développés dans les poumons des fœtus et des nouveau-nés que dans ceux des adultes. Ces cellules sont situées seules ou en groupes dans l'épithélium des bronches et des bronchioles et possèdent une innervation abondante. De nombreuses cellules endocriniennes spécifiques des poumons sont similaires à celles de l’hypophyse, du duodénum, du pancréas et de la thyroïde. Parmi les neuropeptides synthétisés par les poumons, on a trouvé les suivants : leu-enképhaline, calcitonine, polypeptide vaso-intestinal, substance P, etc. Le groupe d'apudocytes le plus nombreux et le mieux organisé du tractus gastro-intestinal sont également les cellules de Kulchitsky (cellules Ec) . Leur fonction est considérée comme la synthèse et l'accumulation d'amines biogènes - sérotonine et mélatonine, ainsi que d'hormones peptidiques - motiline, substance P et catécholamines. De plus, plus de 20 types de cellules (A, D, G, K, etc.) synthétisant des hormones polypeptidiques ont été trouvées dans le tractus gastro-intestinal. Parmi eux figurent l'insuline, le glucagon, la somatostatine, la gastrine, la substance P, la cholécystokinine, la motiline, etc.
Types d'apudopathies. Les troubles de la structure et de la fonction des apudocytes, exprimés par des syndromes cliniques, sont appelés apudopathies. En fonction de leur origine, les apudopathies se distinguent entre les apudopathies primaires (déterminées héréditairement) et secondaires (acquises).
Les apudopathies primaires comprennent notamment le syndrome des tumeurs endocriniennes multiples (MET) de différents types (voir tableau selon N.T. Starkova). Il s'agit d'une maladie autosomique dominante caractérisée par de multiples tumeurs bénignes ou malignes provenant d'apudocytes de diverses localisations. Ainsi, le groupe de maladies appartenant au SMES de type I comprend principalement les patients présentant une forme familiale d'hyperparathyroïdie. Dans ce syndrome, une hyperplasie de toutes les glandes parathyroïdes est détectée en association avec une tumeur du pancréas et (ou) de l'hypophyse, qui peut sécréter un excès de gastrine, d'insuline, de glucagon, de VIP, PRL, STH, ACTH, provoquant le développement de symptômes cliniques correspondants. manifestations. Plusieurs lipomes et carcinomes peuvent être associés à des SMES de type I. L'hyperparathyroïdie est l'endocrinopathie la plus exprimée dans le SMES de type I, et elle est observée chez plus de 95 % des patients. Les gastrinomes (37 %) et les VIPomes (5 %) sont moins fréquents.
Le SMEO de type IIa est caractérisé par la présence chez les patients d'un cancer médullaire de la thyroïde, d'un phéochromocytome et d'une hyperplasie ou d'une tumeur de la glande parathyroïde. La combinaison du cancer médullaire de la thyroïde et du phéochromocytome a été décrite pour la première fois en détail par Sipple (1961), c'est pourquoi cette variante du SMES est appelée syndrome de Sipple.
Des apudopathies secondaires peuvent survenir avec des maladies du système cardiovasculaire ou nerveux, des maladies infectieuses, des intoxications, des tumeurs localisées en dehors du système APUD.
Sur la base de leur prévalence, on distingue les apudopathies multiples (caractérisées par l'implication de différents types d'apudocytes dans le processus pathologique) et les apudopathies solitaires (la fonction de n'importe quel type d'apudocytes est altérée). Un exemple d'une des formes d'apudopathies multiples peut être le syndrome MEO décrit ci-dessus. Parmi les tumeurs solitaires, les plus courantes sont les tumeurs de l'apudom, qui proviennent des cellules du système APUD et ont une activité hormonale. Bien que de telles tumeurs puissent parfois produire plusieurs hormones provenant de différents types de cellules, les manifestations cliniques des apudopathies solitaires sont généralement déterminées par l'action d'une seule hormone. Les apudopathies se distinguent également selon leurs caractéristiques fonctionnelles. Il existe des formes de troubles hyper-, hypo- et dysfonctionnels. La base des deux premières formes est généralement une hyper- ou une hypoplasie des apudocytes, respectivement ; les troubles dysfonctionnels sont caractéristiques des apudopathies multiples. Ci-dessous, nous donnerons une brève description de certaines des hormones peptidiques du système APUD et de leur rôle dans la pathologie.
Gastrine. Ce peptide est produit par les cellules G principalement dans le pylore de l'estomac. Un autre représentant du système APUD a également été identifié : la bombésine, produite par les cellules P, qui est un stimulateur de la libération de gastrine. Par conséquent, la bombésine est appelée hormone de libération de la gastrine. La gastrine est un puissant stimulateur de la sécrétion d'acide chlorhydrique, et cette dernière inhibe sa formation par rétroaction négative. De plus, la gastrine stimule la production d'enzymes pancréatiques, améliore la sécrétion du suc pancréatique et augmente la sécrétion de bile ; inhibe l'absorption du glucose, du sodium et de l'eau dans l'intestin grêle, ainsi qu'une libération accrue de potassium ; stimule l'activité motrice du tractus gastro-intestinal.
En 1955, Zollinger et Ellison ont décrit pour la première fois des patients présentant des ulcères peptiques récurrents, une hypersécrétion sévère d'acide chlorhydrique et une tumeur des cellules des îlots - le gastrinome, produisant une quantité accrue de gastrine. Cette triade de symptômes est appelée syndrome de Zollinger-Ellison. Le gastrinome est le plus souvent localisé dans le pancréas, ainsi que dans la sous-muqueuse du duodénum. Jusqu'à 75 % des gastrinomes pancréatiques et jusqu'à 50 % des gastrinomes duodénaux donnent des métastases. Cliniquement, le syndrome se manifeste par des lésions ulcéreuses à développement rapide (généralement dans le bulbe duodénal), des douleurs épigastriques, des saignements ulcéreux fréquents, des nausées, des vomissements et de la diarrhée.
Glucagon. Une hormone peptidique produite par les cellules alpha des îlots pancréatiques. Le glucagon de poids moléculaire légèrement plus élevé est sécrété par les cellules de la muqueuse duodénale. Le glucagon pancréatique a un effet hyperglycémique prononcé en raison d'une forte augmentation de la glycogénolyse dans le foie sous son influence. L'hormone entérale a un effet stimulant sur la sécrétion d'insuline. Ainsi, le glucagon participe à la stabilisation de la glycémie. Lorsque la glycémie diminue, du glucagon est libéré. De plus, c’est une hormone lipolytique qui mobilise les acides gras du tissu adipeux.
Plus de 100 glucagénomes ont été décrits - des tumeurs malignes hormonalement actives localisées principalement dans la queue du pancréas. Le glucagénome conduit au développement du syndrome de dermatite diabétique. Elle se caractérise par des signes de diabète sucré modéré (dus à une hyperglucagonémie) et des modifications cutanées sous forme d'érythème nécrolytique migrateur. Une glossite, une stomatite, une anémie et une perte de poids se développent également. Les enfants ont souvent des convulsions, des périodes d'apnée et parfois un coma.
Une autre hormone du système APUD est somatostatine(ou libérant de la somatotropine). Cette hormone inhibitrice est produite non seulement dans le système nerveux central (dans l'hypothalamus), mais également dans les cellules D de l'estomac, des intestins et du pancréas, ainsi qu'en petites quantités dans tous les tissus du corps. En plus du rôle physiologique principal - inhibition de la libération de l'hormone somatotrope, la somatostatine inhibe la libération d'insuline, de thyroxine, de corticostérone, de testostérone, de prolactine, de glucagon, ainsi que de gastrine, de cholécystokinine, de pepsine, etc. la somatostatine inhibe l'activité motrice du tractus gastro-intestinal, a un effet sédatif et a la capacité de se lier aux récepteurs opiacés du cerveau, affectant ainsi les mouvements involontaires. De ce qui précède, il s'ensuit que cette hormone joue un rôle très important dans la vie du corps.
Les manifestations cliniques de l'hypersomatostatinémie (avec tumeurs pancréatiques sécrétant cette hormone - les somatostatinomes) sont très polymorphes. Il s'agit de diverses combinaisons de diabète sucré, de lithiase biliaire, d'insuffisance pancréatique exocrine, d'hypo- et d'achlorhydrie gastriques, d'anémie ferriprive, etc.
Polypeptide intestinal vasoactif(VIP). Ce peptide a d'abord été isolé de l'intestin grêle, puis retrouvé dans les formations nerveuses de l'ensemble du tractus gastro-intestinal, ainsi que dans le système nerveux central, les poumons et d'autres organes. VIP inhibe la sécrétion gastrique, active la sécrétion du suc intestinal, ainsi que la sécrétion d'eau et de bicarbonate par le pancréas, provoque un relâchement du sphincter inférieur de l'œsophage et du côlon. De plus, le VIP est capable de provoquer une vasodilatation, une expansion des bronchioles et de stimuler la libération d'hormones par le pancréas et l'hypophyse antérieure ; activer la glucogenèse et la glycogénolyse. Une augmentation de la formation de VIP est le plus souvent observée avec VIPoma - une tumeur endocrine de l'appareil des îlots du pancréas. Cette tumeur conduit au développement du syndrome de Wermer-Morrison, se manifestant par de la diarrhée, de la stéatorrhée, de la déshydratation, une perte de poids, une hypo- et une achlorhydrie. Une hypokaliémie, une hypercalcémie, une acidose et une hyperglycémie se développent. Des convulsions et une hypotension artérielle peuvent survenir. La formation excessive de VIP est la principale cause de diarrhée abondante dans le syndrome de Werner-Morrison (choléra endocrinien).
Et enfin, nous caractériserons un autre peptide du système APUD. Ce substance-R. Il est largement distribué dans le système nerveux central, notamment dans l’hypothalamus, la moelle épinière et les poumons. Dans le tractus gastro-intestinal, la substance P se trouve dans les plexus de Meissner et d'Auerbach, dans les muscles circulatoires et longitudinaux de l'intestin. Dans le système nerveux central, ce peptide joue le rôle d'un neurotransmetteur typique ; il est capable d'accélérer le métabolisme des amines biogènes dans le cerveau et de moduler la réponse à la douleur. Au niveau du tractus gastro-intestinal, il a été établi que la substance P améliore la sécrétion, mais inhibe l'absorption des électrolytes et de l'eau dans l'intestin grêle et provoque la contraction des muscles lisses des organes internes.
Pour conclure la discussion sur le sujet, je voudrais souligner ce qui suit : 1) le matériel présenté indique qu'une organisation structurelle très complexe de la régulation neuroendocrinienne de l'activité vitale s'est développée dans le corps au cours de la phylogenèse et qu'un très large éventail de causes possibles et mécanismes de développement de troubles endocriniens; 2) on peut noter que ces dernières années, notre compréhension de l'étiopathogénie des endocrinopathies s'est considérablement élargie et approfondie. Le sujet d'étude n'était pas seulement la pathologie « classique » du système endocrinien, mais aussi ses types « non classiques ».
1. APUD-SYSTÈME ET SES BASES MORPHOLOGIQUES
L'hypothèse de la présence dans la muqueuse du tractus gastro-intestinal de cellules remplissant une fonction endocrinienne a été formulée dès 1914 par P. Masson. Les travaux de A. Pierce (1968-1976) ont joué un rôle majeur dans l'élaboration de la doctrine de cette fonction du tube digestif. Selon lui, il existe des cellules uniques caractérisées par des points communs embryonnaires, certaines propriétés morphologiques et biochimiques, qui constituent un système APUD (Amine Precursor Uptake Decarboxylation) unique.
Ces cellules se caractérisent par une teneur élevée en amines (Amine). la capacité d'assimiler les précurseurs aminés (Precursor Uptake) et la présence de l'enzyme décarboxylase (Décarboxylation).
Les cellules APUD sont localisées dans l'hypothalamus, l'hypophyse, la glande thyroïde, la médullosurrénale et le tube digestif. Comme l'ont noté K. Welbourn et al. (1974) « Le tube digestif est la plus grande usine endocrinienne du corps. »
Les cellules APUD comprennent 36 types de cellules, dont 28 sont des dérivés de l'ectoderme (A. Pearse et al., 1976), la source des 18 variétés restantes n'a pas encore été clarifiée.
Le nombre de cellules ayant des fonctions non identifiées sur la base des données de coloration et de microscopie électronique liées au système APUD, ainsi que des hormones d'origine inconnue, comme l'ont noté M. Grossman et al (1974) et A. Pearse (1974), est toujours. assez significatif.
L'ensemble du système de cellules APUD est divisé en 3 groupes (A. Pearse, I. Polak. 1978) : 1. Cellules neuroendocrines dérivées de la crête neurale (il existe 7 types, par exemple les cellules C qui produisent du calcium).
2. Cellules provenant de l'ectoderme neutre (il en existe 20 types). Ils sont majoritairement localisés dans les tissus cérébraux, produisant par exemple de la lulibérine, de la thyréolibérine, etc.
3. Cellules du système gastro-intestinal-pancréatique (GEP-celes). Ils sont d'origine ectoblastique. Il s'agit du plus grand groupe de cellules du système APUD.
Hormones du tractus gastro-intestinal et lieux de leur formation
|
Nom de l'hormone |
Site de production d'hormones |
Types de cellules endocriniennes |
|
Somatostatine |
Estomac, intestin grêle proximal, pancréas |
|
|
Peptide intestinal vasoactif (VIP) |
Dans toutes les parties du tractus gastro-intestinal |
Cellules di |
|
Polypeptide pancréatique (PP) |
Pancréas |
|
|
Antre de l'estomac, pancréas, intestin grêle proximal |
||
|
Antre de l'estomac |
||
|
Bulbogastron |
Antre de l'estomac |
|
|
Duocrinine |
Antre de l'estomac |
|
|
Bombésie |
Estomac et intestin grêle proximal |
|
|
Sécrétine |
Intestin grêle |
|
|
Cholécystokinine-pancréozymine (CCK-PZ) |
Intestin grêle |
|
|
Entéroglucagon |
Intestin grêle |
|
|
Intestin grêle proximal |
CE;-cellules |
|
|
Peptide gastroinhibiteur (GIP) |
Intestin grêle |
|
|
Neurotensine |
Intestin grêle distal |
|
|
Enképhalines (endorphines) |
Intestin grêle proximal et pancréas |
|
|
glande |
||
|
Substance P |
Intestin grêle |
UE 1 cellule |
|
Willikinine |
Duodénum |
Cellules EC i |
|
Entérogastron |
Duodénum |
Cellules i de l'UE |
|
Sérotonine |
Tube digestif |
UE]. Cellules ECG |
|
Pancréas |
||
|
Glucagon |
Pancréas |
Les cellules endocrines du tractus gastro-intestinal se caractérisent par les caractéristiques suivantes qui les distinguent des cellules intestinales (entérocytes) :
1. Faible niveau de réticulum endoplasmique granulaire.
2. Teneur élevée en ribosomes libres.
3. Niveau élevé de réticulum lisse sous forme de vésicules.
4. Électrons denses et labiles lorsqu’ils sont fixés par les mitochondries.
5. Vésicules sécrétoires liées à la membrane contenant de l'oxynophylène
nous sommes.
Selon la terminologie unifiée développée, appelée Wiesbaden (1970), avec de nouvelles modifications apportées lors d'une réunion de cinq groupes de recherche (dont des participants à l'Accord de Wiesbaden et un groupe de scientifiques japonais) à Bologne (1973), les types de cellules endocriniennes suivants sont classés dans le tractus gastro-intestinal :
Dans l'estomac - EC, G, ECL, AL, D, D,.
Dans l'intestin - EC, S, EG, G, I, D, D,.
Dans le pancréas - A, B, D, Di.
g-cellules. Grâce à des analyses immunomorphologiques et immunofluorescentes, à l'aide de sérum antigastrine, le lien de ce type de cellules avec la production de l'hormone gastrine a été prouvé. Ces cellules sont localisées dans la muqueuse de la région pylorique de l'estomac, dans ses parties cardiaque et antrale, dans le duodénum, notamment dans son bulbe, et dans le jéjunum (en moindre quantité). La membrane apicale des cellules G contient des microvillosités.
Cellules EC. Des cellules de ce type (cellules d'argentoffine, entérochromaffines, cellules de Kulchitsky) se trouvent tout au long du tractus gastro-intestinal, localisées principalement à la base des glandes pyloriques de l'estomac ou dans la région cryptale des villosités de l'intestin grêle. Les cellules sont équipées de petites microvillosités. Les cellules EC sont productrices de 5-hydroxytryptamine. Cependant, les résultats de recherche obtenus ces dernières années suggèrent qu'en plus de cette substance, les cellules EC produisent un produit polypeptidique, la motiline.
Dans le fond de l'estomac, on trouve des cellules ECL de type entérochromaffine, qui diffèrent des cellules EC par certains détails ultrastructuraux.
PAR EXEMPLE.-cellules(entéroglucagon). Localisé dans la membrane muqueuse de l'intestin grêle et du gros intestin. Les cellules de ce type sont productrices d'entéroglucagon.
1-cellules. Trouvé dans la membrane muqueuse du duodénum et du jéjunum. Leurs granules sont similaires aux granules des cellules EG et S en termes de densité électronique, mais occupent une position intermédiaire en taille (cela a déterminé le nom des cellules - intermédiaire). Les cellules I sont productrices de cholécystokinine-pancréozymine.
S-cellules. Ils sont situés dans les cryptes du duodénum et dans les parties proximales du jéjunum. Chez l'homme, leur nombre est relativement faible. Les cellules S sont productrices de sécrétine.
D-cellules. Ils sont situés dans la membrane muqueuse des parties fundique et pylorique de l'estomac et du jéjunum. Les cellules de ce type synthétisent la somatostatine.
Les cellules du système APUD sont des cellules neuroendocrines hormonalement actives qui ont des propriétés universelles pour absorber les précurseurs d'amines, les décarboxyler et synthétiser les amines nécessaires à la construction et au fonctionnement des peptides régulateurs - les cellules d'absorption et de décarboxylation des précurseurs d'amines (APUD). Les tumeurs du système APUD peuvent être bénignes (apudomes) ou malignes (apudoblastomes).
Caractéristiques générales des tumeurs du système APUD
|
Origine |
Effet hormonal primaire |
Symptômes cliniques caractéristiques |
Prévalence |
Malignité, % |
|
|
Gastrinome |
hypersécrétion d'acide chlorhydrique |
ulcères gastroduodénaux multiples réfractaires au traitement, diarrhée, stéatorrhée |
|||
|
Somatostatinome |
inhibition de la sécrétion d'insuline, de gastrine, de sérotonine, de polypeptide pcréatique |
diabète sucré, diarrhée, stéatorrhée, calculs des voies biliaires, angstrinemie, perte de poids |
|||
|
Glucagonome |
action glycogénolytique et lipolytique |
diabète sucré, éruptions cutanées, thrombose veineuse, anémie, diarrhée, perte de poids |
|||
|
Cellules D1 |
sécrétion massive de liquide et d'électrolytes par l'intestin grêle |
diarrhée aqueuse sévère, hyperkaliémie, hypochlorhydrie, déshydratation, perte de poids |
|||
|
Insulinome |
hypoglycémie avec augmentation des taux d'insuline |
crises d'hypoglycémie |
|||
|
Carcinoïde |
cellules entérochromaffines |
hyperproduction d'insuline, augmentation de la motilité |
rougeur du visage et du corps, diarrhée, bronchoconstriction, fibrose endocardique droite |
Gastrinome. En 1955, lors d'une réunion de l'American Surgical Association, H. Zollinger et E. Ellison ont rendu compte de deux patients présentant des ulcères peptiques duodénaux récurrents, une hypersécrétion sévère d'acide chlorhydrique et une tumeur des cellules des îlots. Cette triade de symptômes est devenue plus tard connue sous le nom de syndrome de Zollenger-Ellison. On suppose que le gastrinome est une tumeur des cellules G du pancréas, préservées depuis le développement embryonnaire - les insulinocytes acidophiles - les cellules. Les cellules tumorales produisent de la gastrine, qui stimule la sécrétion d'acide chlorhydrique par les cellules pariétales situées dans le corps et le fond de l'estomac. Dans la muqueuse gastrique d'une personne en bonne santé, il existe des cellules G qui produisent de la gastrine, dont l'hyperplasie et l'hyperfonctionnement peuvent conduire à des manifestations du syndrome de Zollenger-Ellison.
Chez 80 % des patients, les gastrinomes sont localisés dans le pancréas, chez 15 % - dans la paroi du duodénum, chez 5 % - en dehors de l'intestin (estomac, foie, rate). Une croissance tumorale multifocale est observée dans 60 % des cas. Les tumeurs peuvent être petites et dans 50 % des cas ne sont pas détectées lors de la chirurgie. Dans près de 40 % des cas, les gastrinomes ont métastasé au moment où le diagnostic est posé. Parmi 1 000 patients atteints d'ulcères duodénaux, un souffre de gastrinome. Le gastrinome survient plus souvent chez les hommes.
Dans la plupart des cas, des ulcères duodénaux sont détectés, moins souvent la localisation gastrique des ulcères est déterminée.
Caractéristiques des ulcères du syndrome de Zollenger-Ellison : multiples, résistants au traitement, associés à diarrhée, postbulbaire, récidivante après traitement chirurgical, avec des antécédents familiaux, associée à une hypercalcémie (possible NEM-1), une sécrétion basale supérieure à 15 mmol/h ou 5 mmol/h après résection partielle de l'estomac, des signes radiologiques ou endoscopiques d'hypertrophie de les plis de la muqueuse gastrique.
Le signe diagnostique en laboratoire le plus important du gastrinome est une augmentation du taux de gastrine dans le sérum sanguin lors de la détermination radioimmunologique. La teneur en gastrine dans le syndrome de Zollenger-Ellison augmente jusqu'à 200-10 000 ng/l (la normale est inférieure à 150 ng/l). Si la teneur en gastrine n'est pas clairement augmentée, un test de provocation avec administration intraveineuse de calcium (5 mg/kg par heure pendant 3 heures) ou de sécrétine (3 unités/kg par heure) est utilisé pour différencier le gastrinome et l'hyperfonctionnement des cellules G gastriques. . Le test est considéré comme positif si la teneur en gastrine dans le sérum sanguin augmente de 2 à 3 fois par rapport au niveau basal. Avec les ulcères duodénaux ordinaires, après l'administration de sécrétine, au contraire, il y a une légère diminution des taux de gastrine, et après le gluconate de calcium, l'augmentation des taux de gastrine est insignifiante. L'utilisation du test avec des aliments standardisés (30 g de protéines, 20 g de graisses et 25 g de glucides) ne modifie pas la concentration initiale de gastrine chez les patients atteints de gastrinome, tandis que chez les patients présentant des ulcères ordinaires, une augmentation de sa concentration est observée. .
Il est possible d'établir la localisation exacte du gastrinome par échographie, résonance magnétique nucléaire et tomodensitométrie chez environ 15 à 30 % des patients présentant une taille de tumeur allant jusqu'à 1 cm et chez 80 à 90 % des patients présentant une taille de tumeur supérieure à 1 cm. 2 cm. Les métastases se produisent généralement dans le foie.
Traitement du gastrinome : chirurgical (ablation radicale de la tumeur, et si impossible, gastrectomie totale) ou conservateur (anti-H 2 à fortes doses). Le taux de survie à cinq ans après le diagnostic (même en présence de métastases hépatiques) est de 50 à 80 %. Le taux de survie sur cinq ans pour les opérations radicales atteint 70 à 80 %. La mort survient généralement à la suite de complications liées aux ulcères.
En cas de gastrinome malin et de présence de métastases, la chimiothérapie est réalisée avec de la streptozotocine et du 5-fluorouracile ; ces dernières années, un complément très efficace est l'utilisation de la somatostatine (octréotide), qui inhibe non seulement la formation de gastrine et d'acide chlorhydrique. , mais favorise également la régression de la tumeur et de ses métastases.
Carcinoïde- la tumeur la plus courante du système APUD. Les tumeurs carcinoïdes proviennent de cellules entérochromatophytes et peuvent survenir dans presque tous les organes, mais se trouvent le plus souvent dans le tractus gastro-intestinal. Les carcinoïdes représentent environ 5 % de toutes les tumeurs gastro-intestinales. Localisation de la tumeur : dans 55 % - dans l'appendice, dans 30 % - dans l'intestin grêle, dans 5 % - dans l'estomac, dans 3 % - dans le côlon, 7 % - dans d'autres organes (pancréas, bronches, etc. .). Selon l'origine embryonnaire de la tumeur, la production de sérotonine est plus ou moins possible.
Le tableau clinique est dû à syndrome carcinoïde, qui comprend les composants suivants :
bouffées de chaleur au visage, au cou, à la poitrine
bronchospasme
maladie cardiaque du côté droit.
Les manifestations du syndrome carcinoïde dépendent de la production de sérotonine. Il peut y avoir des tumeurs asymptomatiques, par exemple lors d'une appendicectomie. Les métastases du carcinoïde s'accompagnent du développement du syndrome carcinoïde.
Une méthode de diagnostic en laboratoire consiste à déterminer la concentration accrue d'acide 5-hydroxyindoléacétique (un métabolite de la sérotonine) dans l'urine.
La vérification du diagnostic est histologique lors de l'examen d'un prélèvement chirurgical ou d'une biopsie.
Le traitement est conservateur et chirurgical.
Les patients ne doivent pas consommer d'aliments contenant beaucoup de sérotonine : bananes, ananas, kiwi, noix, etc. Des antagonistes de la sérotonine sont utilisés : cyproheptadine (Peractin) 4 mg 3 à 4 fois par jour, ou méthysergide (Sansert) en dose initiale de 2 mg 3 à 4 fois par jour. L'octréotide (Sandostatine) pour soulager le syndrome carcinoïde est administré par voie sous-cutanée à une dose quotidienne de 0,2 à 0,6 mg (0,1 à 0,2 mg 2 à 3 fois par jour). L'inefficacité de ce traitement, l'excrétion urinaire de plus de 150 mg par jour d'acide 5-hydroxyindoleacétique ou le développement d'une cardiopathie carcinoïde est une indication de chimiothérapie à base de streptozotocine (500 mg pour 1 m2 de surface corporelle) et de 5-fluorouracile (40 mg pour 1 m2) pendant 5 jours. Les cycles sont répétés à intervalles de 6 semaines.
Les tumeurs de moins de 1 cm de diamètre peuvent être retirées localement. Toute la zone touchée est radicalement enlevée (hémicolectomie, gastrectomie subtotale), généralement lorsque le diamètre du carcinoïde est supérieur à 2 cm. En présence de métastases dans le foie, une résection du segment affecté ou une énucléation du ganglion tumoral est réalisée. Après une intervention chirurgicale pour un carcinoïde carcinoïde, une crise carcinoïde peut se développer, accompagnée d'une insuffisance cardiovasculaire, d'une parésie gastrique et intestinale et d'autres symptômes. Une crise carcinoïde peut être stoppée avec succès par l'administration intraveineuse de 0,1 à 0,5 mg de sandostatine.
Tumeurs endocriniennes multiples (MEN)
MEN-1 (syndrome de Wermer) : tumeurs bénignes de l'hypophyse antérieure, hyperplasie (adénome) des glandes parathyroïdes, tumeurs bénignes et malignes multiples des cellules des îlots pancréatiques, tumeurs carcinoïdes. Cliniquement caractérisé par une combinaison de signes d'hyperparathyroïdie et de syndrome de Zollenger-Ellison. Dans 2/3 des cas, la maladie est asymptomatique ; le diagnostic repose sur les résultats d'une étude biochimique (hypercalcémie sur fond de taux de phosphore faibles ou normaux, augmentation des taux d'hormone parathyroïdienne) et sur la présence de complications dues à une hypercalcémie. (lithiase urinaire, néphrocalcinose, lésions osseuses). Le traitement commence par une paratectomie.
MEN-2 (syndrome de Sippile) : carcinome médullaire de la thyroïde, phéochromocytome, hyperplasie (adénome) de la glande parathyroïde.
MEN-3 (syndrome de Gorlin) : carcinome médullaire de la thyroïde, phéochromocytome, neuromatose diffuse multiple des muqueuses, structure corporelle « marfanoïde ».
CHAPITRE 23. CONCEPT DU SYSTÈME APUD ET APUDOMAS. SYNDROME CARCINOÏDECHAPITRE 23. CONCEPT DU SYSTÈME APUD ET APUDOMAS. SYNDROME CARCINOÏDE
Le terme APUD (abréviation des mots anglais : Amine - amine, Precursor - prédécesseur, Uptake - absorption, utilisation, Decarboxylation - décarboxylation) a été proposé par H.G.E. Pearse en 1966 pour indiquer les propriétés générales de diverses cellules neuroendocrines. La collection de ces cellules s’appelait le système APUD. Toutes les cellules du système APUD sont capables d'accumuler du tryptophane, de l'histidine et de la tyrosine et de les convertir par décarboxylation en médiateurs - sérotonine, histamine et dopamine. De plus, toute cellule du système APUD est potentiellement capable de synthétiser de nombreuses hormones peptidiques.
Un certain nombre de tumeurs se développent dans la région de la tête et du cou, affectant considérablement le statut hormonal d’une personne. Ces tumeurs comprennent les tumeurs du système paraganglien et de la glande thyroïde. Fonctionnellement et structurellement, ces tumeurs sont proches des cellules de la médullosurrénale. Le cancer médullaire de la thyroïde sécrète de la calcitonine et des prostaglandines. Des taux élevés de calcitonine ne sont pas cliniquement apparents, mais des taux élevés de prostaglandines provoquent souvent de la diarrhée. Le cancer médullaire de la thyroïde est souvent une composante des NEM de types IIa et IIb (voir chapitre « Tumeurs héréditaires »).
Le phéochromocytome peut être une composante des MEN de types IIa et IIb. Il s’agit généralement d’une tumeur bénigne, hautement différenciée, qui ne métastase pas.
La plupart des cellules du système APUD proviennent de la crête neurale. De nombreuses cellules endodermiques et mésenchymateuses peuvent acquérir les propriétés des cellules du système APUD sous l'influence de stimuli externes. La localisation des organes du système APUD, cellules capables de se transformer en apudoms similaires (d'où des sources possibles d'apudoms), est très diversifiée. Ceux-ci comprennent les organes neuroendocriniens centraux et périphériques (hypothalamus, hypophyse, médullosurrénale, paraganglions), les cellules gliales et les neuroblastes des organes neuroendocriniens centraux et périphériques.
système nerveux. Cellules C de la glande thyroïde, des glandes parathyroïdes, des îlots pancréatiques, des cellules endocrines uniques dans les parois des canaux pancréatiques, des cellules entérochromaffines de la muqueuse gastrique et des cellules neuroendocrines des poumons, ainsi que des mélanocytes de la peau et des muqueuses.
Les cellules des tissus et des tumeurs d'origine contiennent des granules de minoamine, ce qui est particulièrement clairement visible sur les photographies au microscope électronique. Il est maintenant établi que les écarts dans la chimie sanguine stimulent l'activation de l'activité sécrétoire des paraganglions et la libération de monoamines dans le sang, et éventuellement d'autres médiateurs qui régulent l'homéostasie. Le contenu des granules est constitué de catécholamines, de sérotonine et de dopamine. Les médiateurs et hormones sécrétés par les cellules du système APUD régulent le métabolisme des glucides, du calcium et des électrolytes, le tonus vasculaire et musculaire, la sécrétion et l'absorption dans le tractus gastro-intestinal et les poumons, la différenciation et la prolifération des différents types cellulaires. Les médiateurs et les hormones ne sont pas sécrétés en permanence, mais en réponse à des stimuli externes. Au cours de la transformation tumorale des cellules, la sécrétion devient dérégulée et la nature des substances produites peut changer considérablement. De plus, la tumeur primitive et ses métastases peuvent sécréter divers médiateurs et hormones.
Les tumeurs du système APUD, qui se développent généralement lentement et manifestent principalement un effet de type hormonal sur divers organes et systèmes, quelle que soit leur localisation dans les organes internes, sont appelées carcinoïdes. Les carcinoïdes se développent n'importe où dans le tractus gastro-intestinal, y compris l'œsophage, l'estomac, le duodénum, l'intestin grêle, l'appendice, le côlon, le rectum, les voies biliaires, le pancréas et le foie. De plus, des carcinoïdes peuvent apparaître dans le diverticule de Meckel, le larynx, le thymus, les poumons, les seins, les testicules, les ovaires et l'urètre. Les patients atteints de ces tumeurs développent syndrome carcinoïde. Les carcinoïdes sécrètent principalement la sérotonine. La bradykinine, le 5-hydroxytryptophane, les prostaglandines et l'histamine sont sécrétés en plus petites quantités.
La triade classique des signes du syndrome carcinoïde :
UN) bouffées de chaleur et hyperémie, causée par la libération périodique de grandes quantités de bradykinine et de prostaglandines.
b) diarrhée causée principalement par un excès de sérotonine, dans une moindre mesure par un excès de prostaglandines et de bradykinine ;
chat dommages aux valvules cardiaques Le plus souvent, on observe une insuffisance tricuspide (les valvules sont constamment légèrement ouvertes), moins souvent une sténose de la valvule tricuspide ; les lésions valvulaires sont causées par leur fibrose (effet direct de la sérotonine).
Carcinoïde hormonalement inactif. Il n'y a aucune manifestation du syndrome carcinoïde. Les symptômes sont dus à l'effet direct de la tumeur sur le tractus gastro-intestinal et comprennent des douleurs abdominales, une sensibilité, des nausées, des malaises, une perte de poids, une obstruction intestinale, une obstruction des voies biliaires et des saignements gastro-intestinaux. Le diagnostic repose sur l'endoscopie, la radiographie ou la tomodensitométrie, ainsi que la biopsie et l'examen histologique.
Carcinoïde hormonalement actif. Chez les patients atteints du syndrome carcinoïde, l'excrétion quotidienne du métabolite sérotonine est mesurée.
Traitement.Le traitement radical consiste à éliminer la tumeur primitive et, si possible, les métastases au foie et aux ganglions lymphatiques affectés. Cette approche est appropriée car les carcinoïdes et leurs métastases se développent lentement. Si les métastases ne peuvent être éliminées, un traitement palliatif par somatostatine peut être prescrit.