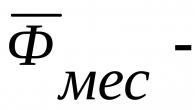Fréquence: trouvé chez environ 1% de la population, diagnostiqué prénatal chez environ 0,2% des enfants.
Attention: 30% des diagnostics prénatals ne sont pas confirmés, un suivi après la naissance est donc nécessaire.
Formes:
- Hydronéphrose : perturbation de la vidange de la vessie (par exemple, valvule urétrale).
- Polykystose rénale.
- Mégalométrie congénitale, duplication.
- Reflux vésico-urétéral.
- Agénésie/aplasie des reins.
- Syndrome de Prune-Belly (syn. : syndrome d'aplasie des membranes de la paroi abdominale, syndrome du « ventre de prune »),
- Exstrophie vésicale.
Complications et problèmes: une uropathie obstructive intra-utérine sévère et prolongée entraîne un rétrécissement du rein (parfois déjà in utero).
PREMIERS SECOURS si vous soupçonnez un défaut du système urinaire :
- Diagnostic postnatal forcé.
- Comptage de la diurèse et collecte des urines. L'urine est excrétée dans les 24 premières heures chez 92 % des nouveau-nés matures et 90 % des nouveau-nés immatures, et dans les 48 heures - chez 99 %.
- Avec la valve, si nécessaire, une ponction sus-pubienne de la vessie est réalisée (déchargement de la vessie et des reins).
Attention: du côté des reins, il n'y a pratiquement pas de conditions d'urgence en salle d'accouchement, mais il peut y avoir une hypoplasie pulmonaire (avec oligo- ou ahydramnios). En cas de maladie rénale, un pneumothorax spontané peut survenir même en cas de respiration spontanée.
PREMIERS SECOURS avec exstrophie vésicale :
- Placer immédiatement sur une couche stérile.
- Hydrater les parties événementielles.
- Évaluation constante de la vessie événementielle : couleur, hémorragie, gonflement.
- On termine le thé prévu ! dans un sac en plastique stérile ou humidifié avec de la vaseline stérile.
- N'oubliez pas de peser votre bébé.
- Chirurgie d’urgence (jusqu’à 24 à 48 heures).
- Examen des voies urinaires proximales.
Hypospadias
Définition : une malformation du pénis avec développement incomplet de l'urètre et une ouverture anormalement localisée sur la partie inférieure du pénis, sur le scrotum ou sur le périnée. Malformation de l'urètre spongieux avec ou sans courbure, défaut ventral du prépuce et hypertrophie du prépuce dorsal.
La tactique dépend de l'emplacement de l'ouverture extérieure :
- Formes capitées, tiges, scrotales, périnéales.
- Avec ou sans sténose.
- Avec ou sans déformation de la tête.
- « Hypospadias sans hypospadias » est une courbure sans luxation significative du foramen.
Combiné avec des vices :
- Cryptorchidie et hernie inguinale - environ 9% des cas.
- Utriculus prostaticus masculinus - environ 11 % des cas (dans les formes sévères).
- Anomalies des reins et du système urinaire - 3%.
Diagnostic différentiel avec intersexualité, hypogénitalisme ; si nécessaire, déterminer le caryotype.
Traitement:
- La correction est indiquée dans tous les cas de courbure du tronc, de sténose du foramen et de localisation du foramen à proximité du sillon coronaire.
- Dans les formes distales, les indications sont déterminées par des troubles esthétiques, mais pas en période néonatale !
Hydrocèle (hydropisie)
Hydrocèle (hydropisie) : La persistance d'un processus vaginal persistant peut conduire à l'accumulation de liquide péritonéal dans le processus vaginalis.
Définition : un kyste rempli de liquide au niveau des membranes testiculaires ou du cordon spermatique.
Formes:
- Hydrocèle du cordon.
- Hydrocèle du testicule : le testicule n'est le plus souvent pas palpable.
- Hydrocèle inguinoscrotale : le pôle supérieur du kyste atteint la cavité abdominale. Une formation dense est palpée en dedans du ligament inguinal dans le petit bassin. En appuyant sur le scrotum, son volume augmente.
- Avec l'hydropisie communicante (la taille change), il existe un lien avec la cavité abdominale. La clinique et le traitement correspondent à une hernie inguinale.
- Kystes du canal Nuco (chez les filles) : accumulations localisées de liquide au niveau du ligament rond en dehors du canal inguinal.
Clinique:
- Formations asymptomatiques unilatérales ou bilatérales de différentes tailles ressemblant à des tumeurs dans la région testiculaire.
- Souvent trouvé chez les nouveau-nés. Régresse spontanément au cours des 3-4 premiers mois de la vie.
- Elles peuvent apparaître spontanément sans cause évidente en dehors de la période néonatale, et elles peuvent également régresser.
- Après la première année de vie, l’hydropisie survient chez moins de 1 % des garçons.
Diagnostique:
- Examen, palpation.
- La diaphanoscopie ne permet pas de distinguer une hydrocèle d'une hernie étranglée.
Diagnostic différentiel : hernie étranglée, hernie scrotale, varicocèle, torsion testiculaire.
Important: La torsion testiculaire peut également être congénitale ; c'est une indication pour une intervention chirurgicale immédiate.
Traitement:
- L'opération est indiquée au bout de six mois en cas d'hydrocèle persistante.
- Au cours des 3 premiers mois, l'intervention chirurgicale est réalisée uniquement en cas d'hydrocèle de grande taille, d'augmentation rapide de volume et d'hydrocèle inguino-scrotale.
Hernie inguinale
Formé à la suite de la persistance du processus vaginal du péritoine. Une taille de trou suffisamment grande permet aux anses intestinales de pénétrer dans l'appendice.
Définition : des parties de l'intestin émergent de la cavité abdominale par une ouverture congénitale ou acquise (orifice herniaire), entourée par le péritoine pariétal. Le contenu de la hernie est limité aux membranes de la hernie (tissu sous-cutané, peau ou paroi du scrotum). Les hernies inguinales des enfants sont toujours indirectes. Ils se forment le long du canal inguinal (persistance du processus vaginal).
Clinique:
- Le plus souvent, il existe des tumeurs asymptomatiques, molles et réductibles dans l'aine, en face du ligament inguinal (hernie inguinale), qui peuvent atteindre le scrotum (hernie scrotale).
- Lorsqu'elle est étranglée, la hernie devient douloureuse et irréductible.
Diagnostique:
- Différence clinique avec l'hydrocèle : avec une hernie inguinale, le sac herniaire est palpé au-dessus de l'anneau inguinal, mais avec une hydrocèle il ne l'atteint pas. Dans les cas douteux, le traitement est toujours réalisé comme pour une hernie étranglée.
- Echographie.
Diagnostic différentiel : testicule dans le canal inguinal, lymphadénite, hydrocèle du testicule ou de la moelle, torsion testiculaire, varicocèle.
Complications:
- Condition potentiellement mortelle.
- Strangulation intestinale ou péritonite.
- Perte d’un testicule/ovaire ou d’une section d’intestin.
Infraction : dans environ 12 % des cas, mais 70 % dans la 1ère année de vie :
- Clinique : apparition soudaine de la maladie, douleur intense, anxiété, symptômes d'irritation péritonéale.
- Résultats : tumeur élasto-élastique, tendue, sédentaire, inguinale ou inguinoscrotale.
Traitement et indications chirurgicales :
- La réduction d'une hernie inguinale se produit souvent indépendamment.
- La réduction active (sous sédation) est possible au plus tard 6 à 8 heures après le début en l'absence de symptômes de choc.
- Si la réduction échoue, une intervention chirurgicale urgente est indiquée.
- Chirurgie élective après réduction réussie.
- Dans tous les autres cas, la réparation précoce de la hernie est optimale afin que le risque d’anesthésie n’augmente pas.
Hernie ombilicale
Hernie ombilicale : défaut du fascia ombilical recouvert de peau. Une intervention chirurgicale est très rarement nécessaire, car le défaut se résorbe avec le temps (vers 1 an de vie). Les pincements ou les lésions cutanées dus à la pression sont extrêmement rares.
PATHOLOGIE PRÉ- ET PÉRINATALE.
PATHOLOGIE DU PLACENTA.
La pathologie prénatale comprend tous les processus et conditions pathologiques du fœtus depuis la fécondation jusqu'à la naissance. Le fondateur de la doctrine de la pathologie prénatale est allemand. scientifique Schwalbe, dont les travaux remontent au début du XXe siècle. La période allant du 196ème jour de développement aux 7 premiers jours après la naissance est appelée pernatale (« autour de l'accouchement ») et est à son tour divisée en prénatale, intra- et postnatale ou prénatale (la période prénatale est plus large et comprend tout ce qui se passe avant le 196ème jour), pendant l'accouchement et le post-partum, néonatal. Tout développement fœtal peut être divisé en 2 périodes : la progenèse (le temps de maturation des gamètes et des cellules germinales) et la cymatogenèse (la période de développement fœtal depuis la fécondation jusqu'à la naissance est divisée en :
Blastogenèse – jusqu'à 15 jours
Embryogenèse – jusqu'à 75 jours
Fœtogenèse – précoce – jusqu’à 180 jours et tardive – jusqu’à 280 jours
Pendant la période de progenèse, la maturation des cellules germinales - ovules et spermatozoïdes - peuvent être endommagées, associées à la fois à des influences exogènes (rayonnements, substances chimiques) et à des modifications héréditaires des chromosomes ou des génomes. Cela s'accompagne de mutations et de maladies héréditaires, notamment d'anomalies congénitales et d'enzymopathies. Les malformations congénitales sont plus souvent observées chez les enfants dont les parents ont plus de 40-45 ans
Les cymatopathies sont des processus pathologiques survenant au cours de la cymatogenèse. La principale pathologie de cette période est constituée de défauts de développement, représentant 20 % ou plus.
Les malformations congénitales sont des modifications persistantes d'un organe ou de l'organisme tout entier qui vont au-delà des variations de leur structure et s'accompagnent généralement de dysfonctionnements. Les causes des malformations congénitales peuvent être divisées en : endogènes et exogènes.
Les causes endogènes incluent les mutations (on estime que 40 %)
Les mutations peuvent être : génétiques – un changement persistant dans le système moléculaire
structure des gènes
chromosomique - changements dans la structure des chromosomes
génomique – modification du nombre de chromosomes, plus souvent
trisomie.
Avec les mutations génomiques, le fœtus meurt le plus souvent. Le rôle de la mutagenèse naturelle est faible. Les mutations induites jouent un rôle important.
Facteurs mutagènes : rayonnements ionisants
chimie. substances (cytostatiques)
virus (rubéole)
surmaturation des cellules germinales (l'allongement du temps entre le moment de la maturation complète des cellules germinales et la formation d'un zygote provoque un complexe de changements pathologiques dans celles-ci.)
Les causes exogènes comprennent :
Rayonnement en cas d'exposition à des périodes critiques du développement embryonnaire. L'irradiation au cours des 3 premiers mois de la grossesse entraîne des malformations congénitales, principalement du système nerveux.
Les effets mécaniques sont le résultat de fusions amniotiques, qui sont un mécanisme de formation du défaut et non sa cause ;
Facteurs chimiques, substances médicinales (thalidomide anticonvulsivant) ;
Alcool – conduit à une embryofœtopathie alcoolique, puis il y a un retard de poids, de taille, de développement mental, il y a une microcéphalie, un strabisme, une lèvre supérieure fine, des malformations cardiaques fréquentes sous forme de malformations septales
Diabète maternel - embryopathie diabétique avec formation de malformations congénitales, malformations du squelette, malformations cardiaques, système nerveux, poids fœtal élevé, syndrome cushingoïde, hyperplasie des îlots de Langerhans. Il existe des signes d'immaturité, de cardio-hépato-splénomégalie, de microangiopathie, de pneumonie.
Virus (cytomégalie)
Le schéma de cette période est une dysontogenèse avec un impact sur le fœtus. Le moment de l'exposition à un agent tératogène est important : différents agents à la même période de développement fœtal donnent les mêmes malformations congénitales, et le même agent à différents moments donne des malformations différentes.
DÉFAUTS CONGÉNAUX DU DÉVELOPPEMENT.
Malformations congénitales du système nerveux central.
Prenez la 1ère place. L'étiologie est variée. A partir de l'exo, l'influence du virus de la rubéole, de la cytomégalie, de Coxsackie, de la polio, etc. a été précisément établie. Médicaments (quinine, cytostatiques), énergie des radiations, hypoxie, mutations génétiques, b-ni chromosomique.
L'anencéphalie est une agénésie du cerveau dont les parties antérieure, moyenne et postérieure sont absentes. La moelle allongée et la moelle épinière sont préservées. Au niveau du cerveau, la connexion. tissu riche en vaisseaux sanguins.
L'acranie est l'absence des os de la voûte crânienne.
Microcéphalie – hypoplasie du g.m. combinée à une diminution du volume des os de la voûte crânienne et à un épaississement.
La microgyrie est une augmentation du nombre de circonvolutions cérébrales avec une diminution de leur taille.
La porencéphalie est l'apparition de kystes communiquant avec les ventricules latéraux.
Hydrocéphalie congénitale - accumulation excessive de liquide céphalo-rachidien dans les ventricules (internes) ou dans les espaces sous-arachnoïdiens (externes) - atrophie du cerveau due à un écoulement altéré du liquide céphalo-rachidien.
Cyclopie – un ou deux globes oculaires dans une orbite
Hernies du cerveau et de la moelle épinière - méningocèle - présence uniquement de membranes dans le sac herniaire, mérinoencéphale - et du cerveau, myélocèle - hernie de la moelle épinière.
Rasischis est un défaut complet de la paroi postérieure du canal rachidien, des tissus mous de la peau et des méninges et de la moelle épinière.
Malformations congénitales des organes digestifs.
On les retrouve dans 3 à 4 % des autopsies des défunts et représentent 21 % de l'ensemble des malformations congénitales.
Des atrésies et des sténoses sont observées dans l'œsophage, le duodénum, le segment proximal du jéjunum et l'iléon distal, dans le rectum et l'anus. Il peut y avoir des fistules trachéo-œsophagiennes dans l’œsophage, entraînant une pneumonie par aspiration grave. Les atrésies peuvent être uniques ou multiples. Dans la zone d'atrésie, l'intestin a l'apparence d'un cordon tissulaire dense et connecté qui, sous l'influence du péristaltisme, peut s'étirer et se briser.
Duplication de sections individuelles de l'intestin - n'affecte le plus souvent que la membrane muqueuse, la couche musculaire étant commune. La zone dupliquée peut prendre la forme d’un kyste, d’un diverticule ou d’un tube. Le défaut se complique de saignements, d'inflammation, de nécrose avec perforation.
Maladie de Hirschsprung - aganglionose segmentaire mégaoclonie - absence de neurones du plexus intermusculaire de la partie inférieure du sigmoïde et du rectum. En raison de la préservation du plexus sous-muqueux (Meissner), la section aganglionnaire de l'intestin est contractée spastiquement au-dessus de celle-ci, une distension de l'intestin se produit avec le méconium ou les selles, suivie d'une hypertrophie compensatoire de la couche musculaire. Les patients souffrent de constipation, une coprostase et une obstruction se développent.
La sténose hypertrophique du pylore est une hypertrophie congénitale des muscles de la région pylorique avec un rétrécissement de sa lumière. Des vomissements persistants sont observés, de 3 à 4 semaines jusqu'à l'apparition d'un coma dû à une perte de chlorures.
Défauts du tube digestif associés à la préservation de certaines structures embryonnaires. Il s'agit notamment d'une hernie ombilicale - un défaut de la paroi abdominale antérieure avec saillie d'un sac herniaire translucide formé par le cordon ombilical et l'amnios, contenant des anses de l'intestin grêle.
Éventarisation des organes abdominaux avec son hypoplasie - la paroi abdominale est ouverte, le sac herniaire est absent.
Les kystes et les fistules au niveau de l'anneau ombilical sont dus à la persistance du canal vitellin.
Le diverticule de Meckel est une saillie en forme de doigt de la paroi de l'iléon.
Anomalies congénitales du foie et des voies biliaires - foie polykystique, atrésie et sténose des voies biliaires extrahépatiques, agénésie et hypoplasie des voies biliaires intrahépatiques dans le tractus porte dans la zone de la triade, conduisant au développement d'une cirrhose biliaire congénitale, d'une hyperplasie congénitale de les canaux intra-hépatiques.
Malformations congénitales du système génito-urinaire.
L'étiologie n'est pas associée à certains facteurs exogènes, mais à l'hérédité et à la famille, et se produit avec des aberrations chromosomiques.
L'agénésie rénale est l'absence congénitale d'un ou des deux reins, l'hypoplasie est une diminution congénitale de la masse et du volume des reins, unilatérale ou bilatérale, la dysplasie rénale est une hypoplasie avec présence simultanée de tissu embryonnaire dans les reins, les gros reins kystiques sont une hypertrophie des reins avec formation de nombreux petits kystes, fusion des reins (rein en fer à cheval) et dystopie. – ne se manifestent pas cliniquement.
Malformations congénitales des voies urinaires :
Duplication du bassin et de l'uretère
Agénésie, atrésie, sténose urétérale
Mégauretère – dilatation soudaine des uretères
Exstrophie - à la suite de son aplasie de sa voie. parois, péritoine et peau de la région pubienne.
Agénésie vésicale
Atrésie, sténose urétrale, hypospadias - un défaut de la paroi inférieure, épispadias - la paroi supérieure de l'urètre chez les garçons.
Tous les défauts de développement entraînent une altération de l'écoulement de l'urine et, sans traitement chirurgical rapide, conduisent à une insuffisance rénale.
Malformations congénitales du système respiratoire :
Aplasie et hypoplasie des bronches et des poumons
Les kystes pulmonaires, multiples et uniques, conduisent ensuite au développement de bronchectasies
Trachéobronchomalacie - hypoplasie du tissu élastique et musculaire de la trachée et des bronches principales, conduisant à la formation de diverticules ou à une expansion diffuse de la trachée et des bronches
Emphysème congénital dû à une hypoplasie du cartilage. Tissu élastique et musculaire des bronches.
Toutes les malformations pulmonaires congénitales, si elles sont compatibles avec la vie, se compliquent facilement d'une infection secondaire avec développement d'une maladie chronique. bronchite et pneumonie avec développement d'un cœur pulmonaire.
Malformations congénitales du système ostéoarticulaire :
Défauts systémiques :
Chondrodystrophie fœtale et achondroplasie (développement altéré des os d'origine cartilagineuse, les os d'origine du tissu conjonctif se développent normalement - raccourcissement et épaississement des membres
Ostéogenèse imparfaite – fragilité osseuse congénitale
B-n marbré congénital – ostéosclérose sévère avec perturbation simultanée du développement du tissu hématopoïétique
Luxation congénitale et dysplasie de l'articulation de la hanche
Amputation congénitale ou amélie des membres
Phocomélie – sous-développement des membres proximaux
Polydactylie - une augmentation du nombre de doigts
La syndactylie est la fusion des doigts.
Envoyer votre bon travail dans la base de connaissances est simple. Utilisez le formulaire ci-dessous
Les étudiants, étudiants diplômés, jeunes scientifiques qui utilisent la base de connaissances dans leurs études et leur travail vous en seront très reconnaissants.
Posté sur http://www.allbest.ru/
GBOU VPO "ORENBOURGMINISTÈRE DE L'ACADÉMIE MÉDICALE D'ÉTAT
DÉPARTEMENT D'HISTOLOGIE, CYTOLOGIE ET EMBRYOLOGIE
ABSTRAIT
sur le sujet: « Anomalies du développement des organes du système urinaire»
Pardiscipline : Histologie
Orenbourg 2014
Introduction
1. Anomalies du développement rénal
2. Anomalies dans le développement de la vessie
3. Méthodes de diagnostic des défauts du système urinaire
Conclusion
Bibliographie
Introduction
Le système urinaire est un ensemble d’organes qui produisent et sécrètent de l’urine. L'urètre est le tube qui transporte l'urine des zones productives des reins vers la vessie, où elle est stockée puis évacuée par un canal appelé urètre. En cas de développement anormal congénital (anomalies) du système urinaire, la production ou l'excrétion d'urine est altérée.
Anomalies du système urinaire sont appelés troubles du développement des organes du système génito-urinaire qui ne permettent pas au corps de fonctionner normalement.
Les anomalies du système urinaire varient en gravité, allant de mineures à potentiellement mortelles. La plupart sont graves et nécessitent une correction chirurgicale. D'autres défauts n'entraînent pas de dysfonctionnement du système urinaire, mais rendent difficile le contrôle de la miction.
1. Anomalies de développementrein
Aplasie unilatérale (agénésie) du rein- l'absence d'un des reins suite à l'arrêt du développement de l'organe. Souvent, avec ce défaut, il n'y a pas de canal déférent correspondant. L'aplasie ne se manifeste généralement d'aucune façon et est détectée lors d'un examen préventif. Dans ce cas, dans la plupart des cas, l'uretère, l'orifice urétéral et la partie correspondante du triangle vésical sont absents. Cependant, dans 15 % des cas d'agénésie, le tiers inférieur de l'uretère est déterminé du côté atteint. Souvent A.P. associées à d'autres anomalies : monorchidie, atrésie de l'anus et du rectum, malformations de l'intestin grêle et du gros intestin.
L'aplasie s'accompagne d'une augmentation de la charge fonctionnelle sur le rein opposé. L'aplasie bilatérale est une pathologie extrêmement rare et incompatible avec la vie.
Duplication rénale- l'orgue semble être composé de deux parties. En conséquence, la taille du rein doublé est plus grande que la normale. Les moitiés supérieure et inférieure sont séparées par un sillon, qui peut avoir différents degrés de gravité, tandis que la partie supérieure de l'organe est toujours plus petite que la partie inférieure. Les deux parties ont un approvisionnement en sang séparé. Le doublement peut être complet ou non. La différence est qu'en cas de doublement complet, les moitiés supérieure et inférieure du rein ont leur propre système pyélocalicien. Dans le même temps, le système pyélocalicien dans la partie inférieure est formé normalement, mais dans la partie supérieure, il est sous-développé. Un uretère part de chaque bassin - duplication de l'uretère (il peut aussi être complet : les deux uretères s'écoulent indépendamment dans la vessie ; et incomplet : l'uretère supérieur fusionne avec l'uretère inférieur). Le traitement de la pathologie elle-même n'est souvent pas nécessaire. Cependant, un rein double est sujet au développement de divers processus pathologiques (hydronéphrose, pyélonéphrite, tuberculose rénale, lithiase urinaire). Dans ces cas, un traitement adapté à la maladie est effectué, ainsi qu'un traitement visant à éliminer la cause profonde (altération de l'urodynamique, altération du flux sanguin).
Rein accessoire- en fait, il y a 3 organes. Le rein accessoire dispose d'un apport sanguin séparé et de son propre uretère, qui peut se déverser dans la vessie ou dans l'uretère du rein principal. Les dimensions du P. supplémentaire sont toujours plus petites que le principal et peuvent varier considérablement. Le P. supplémentaire est toujours situé en dessous du principal (localisation lombaire, iliaque ou pelvienne). En l’absence de complications, la tactique est prudente. Les indications pour l'élimination de P. supplémentaires sont : l'hydronéphrose, le reflux vésico-urétéral, la néphrolithiase, la pyélonéphrite, la tumeur.
Anomalies de la taille des reins
Hypoplasie rénale- réduction de taille due au sous-développement, mais sans perturbations de la structure et du fonctionnement de l'organe. Le plus souvent, la pathologie est unilatérale. Le traitement de l'hypoplasie rénale n'est nécessaire qu'en présence de complications (pyélonéphrite, hypertension artérielle).
Anomalies de localisation (dystopie) des reins
Au cours du développement intra-utérin, le rein en développement se déplace de la région pelvienne vers la région lombaire. La perturbation de ce processus de mouvement conduit à la formation dystopie rénale. Ce défaut peut être unilatéral ou bilatéral. Souligner:
Dystopie lombaire- le rein est situé plus bas que d'habitude et est palpé dans l'hypocondre. Dans ce cas, la néphroptose est parfois diagnostiquée par erreur. Une caractéristique distinctive de la néphroptose de la dystopie est le fait que pendant le prolapsus, les vaisseaux sont dirigés de haut en bas vers le rein et qu'en cas de dystopie, ils s'étendent perpendiculairement. Rarement, le P. dystopique provoque des douleurs abdominales.
Dystopie iléale- l'organe est situé dans la fosse iliaque, les vaisseaux sont généralement de nature multiple et naissent de l'artère iliaque commune. Le principal symptôme de la dystopie iléale est une douleur dans la région abdominale. Des signes d’obstruction du flux urinaire peuvent également apparaître.
Dystopie pelvienne- l'organe est situé profondément dans le bassin, entre la vessie et le rectum. Cette disposition perturbe le fonctionnement du rectum et de la vessie et s'accompagne de symptômes correspondants.
Dystopie thoracique- L'organe est situé dans la cavité thoracique. Ce défaut est rare et survient principalement à gauche. Cela peut souvent se manifester par des douleurs, surtout après avoir mangé. Habituellement diagnostiqué accidentellement.
Dystopie croisée- l'un des reins se déplace du côté opposé au cours de son développement intra-utérin. Dans ce cas, 2 organes sont identifiés d'un côté à la fois. Rarement vu. Il faut dire aussi que la dystopie croisée s’accompagne parfois de la fusion de deux reins. Si deux reins sont détectés d'un côté à la fois, la néphroptose doit être exclue. Le traitement chirurgical n'est indiqué que lorsque des processus pathologiques secondaires sont identifiés (pyélonéphrite, néphrolithiase, hydronéphrose, tumeur).
Anomalies de relation - fusion des reins
Selon le type de fusion rénale, on distingue plusieurs types de fusion :
Bourgeon de biscuit- fusion des reins le long de la surface médiale.
en forme de S- fusion du pôle supérieur d'un rein avec le pôle inférieur de l'autre.
En forme de L (en forme de bâton)- la fusion du pôle supérieur d'un rein avec le pôle inférieur de l'autre se produit également, mais en même temps le premier rein se déplie, entraînant la formation d'un organe ressemblant à la lettre L.
Fer à cheval- fusion des pôles supérieurs ou inférieurs. En conséquence, l’orgue ressemble à un fer à cheval.
Anomalies du nombre de reins
Aplasie(agénésie) L'uretère est très rare et représente 0,2 % des anomalies rénales et urinaires. L'anomalie bilatérale est généralement associée à une agénésie rénale bilatérale, moins souvent à un rein multikystique bilatéral. Cette anomalie n’a aucune signification clinique car incompatible avec la vie.
L'aplasie urétérale unilatérale est également une composante de l'aplasie rénale et le résultat de l'absence de germe urétéral. Parfois, l'uretère se présente sous la forme d'un mince cordon fibreux ou d'un processus qui se termine aveuglément.
Le diagnostic de l'aplasie urétérale repose sur les données de l'urographie excrétrice, qui permet d'établir l'absence de fonction d'un des reins. La cystoscopie révèle une hypoplasie ou une absence totale de la moitié du triangle vésical. L'ouverture de l'uretère peut être située à sa place habituelle, mais être rétrécie. Avec une observation à long terme, l'absence de ses contractions peut être détectée. Parfois, le trou ressemble à une dépression aveugle, déterminée lors de l'insertion du cathéter, ou se termine aveuglément à n'importe quel niveau. Dans ces cas, la cystographie est très informative. En cas d'ouverture rudimentaire de l'uretère, il est recommandé de réaliser une échographie et une tomodensitométrie.
La nécessité d'un traitement ne se pose que lorsque l'uretère se termine aveuglément, car cette anomalie peut provoquer un processus inflammatoire, parfois accompagné de suppuration (empyème) et de formation de calculs. En cas de cicatrice, l'ouverture périphérique de l'uretère forme une cavité fermée, ressemblant à un kyste ou à une tumeur de la cavité abdominale.
De telles complications se manifestent par des douleurs dans la région inguinale ou épigastrique correspondante, une dysurie, une augmentation intermittente de la température corporelle et des phénomènes d'intoxication chronique. L'urine contient un grand nombre de leucocytes, de protéines et de bactéries. Si un calcul est présent, une macro- ou microhématurie est détectée.
Le traitement consiste à retirer le moignon urétéral.
Doubleruretère- une des anomalies les plus nombreuses (1:140). Elle est causée par la croissance simultanée de deux uretères à partir de deux pousses urétérales du blastème néphrogénique ou par la division d'une seule pousse urétérale. L'un des uretères peut se développer normalement, tandis que l'autre peut se développer pathologiquement. Si plusieurs ébauches urétérales se forment dans la section caudale du canal du rein primaire, non seulement un doublement, mais également un triplement des uretères morphologiquement complets sont possibles. Les deux uretères correspondent à deux bassins rénaux, qui sont des collecteurs d'urine pour différentes extrémités du rein.
Dans de tels cas, les reins sont rarement isolés. Un troisième bourgeon supplémentaire est formé.
Parfois, deux uretères ou plus s'étendent du bassin d'un rein non dupliqué, ou l'extrémité proximale de l'un des uretères se termine aveuglément. Les deux uretères traversent généralement la même gaine fasciale.
Une duplication complète (uretère duplex) et incomplète (fissus de l'uretère) des uretères est observée. En cas de duplication incomplète, les deux uretères s'étendent du bassinet du rein jusqu'à la vessie et fusionnent en un seul à des distances différentes de celle-ci. Dans ce cas, un trou apparaît dans la vessie - un uretère fendu. Parfois, les uretères s'écoulent près de la vessie, par voie intra-vésicale (intra-muros) ou même à l'ouverture. L'un des uretères se jette dans l'autre selon un angle aigu.
En règle générale, la longueur des deux uretères depuis le segment urétéropelvien jusqu'à la confluence est différente et les sections des deux uretères situées au-dessus se trouvent dans des phases de péristaltisme différentes. En cas de doublement incomplet, la division est observée principalement dans le tiers supérieur de l'uretère, moins souvent - au milieu et chez 1/3 des patients - dans le bas.
La duplication de l'uretère, complète ou incomplète, est souvent unilatérale. Localisé des deux côtés avec la même fréquence.
En cas de duplication complète, les deux uretères se dirigent séparément vers la vessie. Ils sont étroitement adjacents aux parois, selon la loi de Weigert-Meyer, ils se croisent dans les sections proximales et distales et s'ouvrent par deux ouvertures sur la moitié correspondante du triangle vésical (l'une au-dessus de l'autre ou l'une à côté de l'autre), si il n'y a pas d'ectopie chez l'un d'eux. Dans la vessie, les orifices urétéraux du bassin supérieur sont presque toujours contenus sous les orifices urétéraux du bassin inférieur. Le reflux vésico-urétéral est plus souvent observé avec une duplication complète des uretères. Cela est dû à la courte section intravésicale de l'uretère, qui s'ouvre plus proximale. Parfois, une terminaison aveugle de l'un des uretères dupliqués est observée. L'anomalie se manifeste par des douleurs dans la région épigastrique et des symptômes du processus inflammatoire.
La duplication des uretères est souvent associée à d'autres malformations : absence des deux ou d'un seul orifice urétéral, rétrécissement (rétrécissement urétéral, urétérocèle, ectopie de l'ouverture d'un des uretères (généralement celui du bas), dysplasie segmentaire ou étendue des voies neuromusculaires). éléments des uretères, vaisseaux aberrants, adhérences, cordons fibreux, etc. .P..
Avec un double uretère, aucun symptôme caractéristique n'est observé. Pendant longtemps, l'anomalie a une évolution asymptomatique. Les manifestations cliniques surviennent lorsque des complications surviennent. Les symptômes sont déterminés par la nature et le stade de la complication ou des anomalies associées.
traitement des reins de la vessie
2. Anomalies du développement de la vessie
Anomalies du canal urinaire. Le canal urinaire, ou ouraque, dans l'embryon humain est un vestige de l'allantoïde et s'atrophie avec la transition du fœtus vers la circulation sanguine placentaire à un stade précoce du développement intra-utérin. Malgré la perte de fonctions (apport d'oxygène au fœtus, substances protéiques, fonction excrétrice), le développement inverse de l'allantoïde n'est pas observé et l'ancien canal urinaire est préservé. Il participe à la formation du cordon ombilical et se transforme avec le temps en ligament ombilical médian. Au moment de la naissance, le canal urinaire est oblitéré et prend l’apparence d’un cordon solide qui s’atrophie progressivement.
Dans certains cas, le canal urinaire reste ouvert sur toute sa longueur (non-fermeture complète – fistule vésico-ombilicale) ou dans certaines zones (fin aveugle de la non-fermeture du tronçon externe – fistule ombilicale ; tronçon interne – diverticule vésical, kyste de l'ouraque).
Dans certains cas, notamment chez les prématurés, l’oblitération des voies urinaires survient au cours de la première année de vie. Mais il existe souvent une non-fermeture partielle de celui-ci, qui ne se manifeste pas cliniquement chez l'adulte.
L'obstruction partielle et complète du canal urinaire est une anomalie, mais on n'y prête attention que lorsque certains symptômes apparaissent. La non-fermeture complète du canal urinaire – fistule vésico-ombilicale – est extrêmement rare. Le plus souvent, il y a non-fermeture d'une de ses sections : ombilical - fistule ombilicale ; milieu - kyste de l'ouraque; kystique - diverticule de la vessie.
Si le canal urinaire est complètement ouvert, l'urine s'écoule du nombril, plus souvent pendant la miction. Dans ce cas, la majeure partie de l'urine est excrétée par l'urètre. Ce rapport dépend du diamètre de la lumière du conduit non fermé. Parfois, l'écoulement de l'urine du nombril s'arrête (gonflement de la membrane muqueuse du canal urinaire, la remplissant de granulations ou de masse protéique), puis reprend. La peau du péri-ombilic est macérée en raison de fuites urinaires constantes. Dans certains cas, il existe un tubercule de tissu de granulation (une section de cordon ombilical préservée).
La non-fermeture de la partie médiane du canal urinaire est également assez rare. Dans ce cas, les deux extrémités du conduit étant oblitérées, une section aveugle du canal subsiste entre elles. L'épithélium tapissant le canal produit du mucus qui, s'accumulant dans le canal, étire ses coudes et leur donne une forme arrondie. Progressivement, un kyste se forme au site de formation, qui est palpé entre le nombril et la vessie sous la forme d'une tumeur inactive ronde ou ovale de différentes tailles.
Agénésie vésicale, c'est-à-dire son absence congénitale, une anomalie extrêmement rare. Il est généralement associé à d’autres défauts de développement et incompatible avec la vie.
Duplication de la vessie observé très rarement. Avec cette anomalie, il existe un septum entre les moitiés droite et gauche de la vessie. L'ouverture de l'uretère s'ouvre dans chaque moitié. Chaque moitié de la vessie possède un col. La duplication complète de la vessie est associée à une duplication de l'urètre.
En cas de duplication incomplète, la vessie a un col commun et un urètre. Très rarement, il existe une cloison complète de la vessie, qui la divise dans le sens sagittal ou frontal.
Le traitement consiste à retirer la cloison vésicale.
Hypertrophie congénitale du muscle qui expulse l'urine (détrusor), arrive très rarement. La perméabilité du segment vésico-urétéral n'est pas altérée. L'anomalie entraîne des lésions des voies urinaires supérieures et des reins en raison de la compression des parties putripésiques des deux uretères.
Le seul traitement consiste à retirer la vessie et à la remplacer par une anse intestinale.
Diverticule vésical- saillie sacculaire de sa paroi. Il peut être unique ou multiple. Le diverticule congénital est causé par une formation anormale de la paroi de la vessie. Le plus souvent, un diverticule se forme près des ouvertures des uretères et dans les parties latérales de la vessie, moins souvent à son sommet ou dans sa partie inférieure. La paroi du diverticule est constituée des mêmes couches que la paroi de la vessie. On observe souvent un diverticule volumineux, dépassant le volume de la vessie.
Les anomalies du développement de l'urètre comprennent :
Duplication de l'urètre.
Diverticule urétral congénital.
L'hypospadias est une fente dans la paroi postérieure de l'urètre. Cette anomalie du développement est plus fréquente chez les garçons.
L'épispadias est une non-fermeture de la paroi dorsale (antérieure) de l'urètre. L'épispadias survient chez 1 nouveau-né sur 50 000 et est cinq fois plus fréquente chez les garçons que chez les filles.
L'obstruction infravésiculaire est une obstruction à l'écoulement de l'urine au niveau du col de la vessie ou de l'urètre. L'obstruction infravésiculaire peut être causée par une contracture congénitale du col de la vessie, des valvules urétrales congénitales, une hypertrophie du tubercule spermatique ou une oblitération congénitale (fusion) de l'urètre.
3. Mméthodes de diagnostic des anomalies du système urinaire
Pour le diagnostic précoce des malformations congénitales, l'échographie joue un rôle important pendant la grossesse. Il permet de déterminer avec précision l'oligoamnios, qui résulte le plus souvent d'une diminution de la quantité d'urine excrétée par le fœtus en l'absence de rein ou d'une stagnation des urines dans les voies urinaires. Si le défaut apparaît avant 28-32 semaines de grossesse, il entraîne un sous-développement des poumons et des défauts dans la structure du corps fœtal. Le diagnostic des kystes rénaux et de la stagnation de l'urine dans les voies urinaires fœtales dépend en grande partie des capacités de l'appareil. En cas de suspicion de malformations des voies urinaires, l'accouchement doit être effectué dans des cliniques où se trouvent un néonatologiste, un néphrologue pédiatrique et un urologue.
Avec une évolution pathologique de la grossesse, notamment des oligohydramnios et des malformations congénitales du système urinaire chez les proches, on peut supposer des malformations du système urinaire chez le fœtus. L’absence de diurèse (miction) au cours des deux premiers jours de la vie d’un nouveau-né, les troubles urinaires et les modifications de la quantité d’urine par rapport à la norme d’âge devraient être des indications pour un examen du système urinaire.
Les anomalies des reins et des voies urinaires sont plus fréquentes chez les enfants présentant d’autres anomalies du développement que dans la population générale. Chez certains enfants, le tout premier examen après la naissance révèle des signes d'hydronéphrose au niveau de la cavité abdominale : hypertrophie importante des reins, de la vessie ou des uretères énormes. Des anomalies de la colonne vertébrale, de la moelle épinière ou l'absence de vertèbres sacrées peuvent indiquer une vessie neurogène.
Le diagnostic d'insuffisance rénale est compliqué par la présence dans certains cas d'une miction normale pendant la période néonatale. S'il y a des troubles de la miction, un effort pour uriner ou un jet d'urine intermittent chez les garçons, on peut suspecter la présence d'une obstruction à l'arrière de l'urètre, et une fuite d'urine en gouttes chez les filles peut indiquer une anomalie de la vessie. Chez certains nouveau-nés, l'hydronéphrose, associée à la présence d'une obstruction des voies urinaires, ne peut être détectée qu'après le deuxième jour, lorsque se produit la « séparation physiologique d'une petite quantité d'urine ». Après détection de la dilatation des voies urinaires à l'échographie, une cystographie est réalisée, qui repose sur l'introduction d'un produit de contraste par l'urètre dans la vessie.
Une radiographie est prise pendant que l'enfant urine. La veille de la cystographie, le jour de l'examen et deux jours après l'examen, l'enfant doit prendre des médicaments qui empêchent le développement d'une infection pénétrant dans la vessie lors du cathétérisme et de l'introduction du produit de contraste (bien que des mesures aseptiques soient utilisées). La combinaison de la cystographie et de l'échographie permet d'examiner l'écoulement de l'urine depuis la vessie et à travers les uretères, ainsi que de prouver la présence d'une obstruction dans la partie postérieure de l'urètre. En cas d'infection des voies urinaires, une cystographie doit être réalisée s'il n'y a aucun changement à l'échographie et seulement après traitement de l'infection.
La scintigraphie isotopique est utile pour évaluer la fonction rénale. L'exposition aux rayonnements pour cette étude est nettement moindre.
Conclusion
Les anomalies de l'appareil génito-urinaire sont les plus courantes de toutes les malformations congénitales. Les malformations congénitales sont des modifications persistantes des tissus ou des organes qui vont au-delà des variations de leur structure. La formation de tels défauts résulte d'une perturbation du déroulement normal du développement intra-utérin de l'embryon. La fréquence des malformations congénitales grossières accompagnées de dysfonctionnements dans la population humaine est de 2 à 3 %.
On pense que le plus souvent, les anomalies du système urinaire sont dues à l'influence de facteurs héréditaires et à divers effets négatifs sur le fœtus au cours du développement intra-utérin. Des anomalies du système urinaire peuvent se développer chez un enfant en raison de la rubéole et de la syphilis dont souffre la mère au cours des premiers mois de la grossesse. L'apparition d'anomalies peut être provoquée par l'alcoolisme et la toxicomanie de la mère, son utilisation de contraceptifs hormonaux pendant la grossesse, ainsi que des médicaments sans prescription médicale.
Bibliographie
1. Markosyan A.A. Questions de physiologie liée à l'âge. - M. : Éducation, 1974
2. Sapin M.R. - ANATOMIE ET PHYSIOLOGIE HUMAINES avec caractéristiques liées à l'âge du corps de l'enfant. - centre d'édition "Académie" 2005
3. Petrishina O.L. - Anatomie, physiologie et hygiène des enfants en âge d'aller à l'école primaire. - M. : Éducation, 1979
4. N.V. Krylova, T.M. Soboleva « L'appareil génito-urinaire », anatomie en schémas et dessins, maison d'édition de l'Université de l'Amitié des Peuples de Russie, Moscou, 1994.
5. "Guide to Urology", édité par N. A. Lopatkin, maison d'édition "Medicine", Moscou, 1998. Téléchargez gratuitement l'intégralité de l'essai "Système urinaire humain"
Sources Internet
1. http://kotikit.ru/qanda/anomalii-mochevydelitelnoj-sistemy/
2. http://hvoroby.ru/systemy/47-anomalii -systemy.html
Publié sur Allbest.ru
...Documents similaires
Fréquence d'apparition des anomalies du système urinaire, leurs types et leurs conséquences. Types d'anomalies du système urinaire. Caractéristiques des malformations congénitales des reins et de la vessie : arène bilatérale, duplication de la vessie, mégacalycose, localisation des uretères.
présentation, ajouté le 12/11/2013
Les principales étapes du développement embryonnaire des organes du système urinaire. Caractéristiques de l'anatomie et anomalies existantes des reins : Anomalies de quantité, hypoplasie, dystopie, fusion. Histologie des voies urinaires chez le nouveau-né, ainsi que chez l'enfant.
présentation, ajouté le 26/04/2016
Description et caractéristiques des anomalies rénales : agénésie, hypoplasie, hyperplasie. Ouverture ectopique de l'uretère, ses causes et ses conséquences. Etude des anomalies dans la structure du système urinaire. L'exstrophie de la vessie est l'absence congénitale de sa paroi antérieure.
présentation, ajouté le 12/08/2014
Classifications des anomalies rénales. Sténose fibromusculaire. Fistules artérioveineuses congénitales. L'aplasie est l'absence congénitale d'un ou des deux reins et vaisseaux rénaux. Diverticule du calice ou du bassin. Le rein double. Anomalies des uretères et de la vessie.
présentation, ajouté le 16/07/2017
Classification des maladies du système urinaire. La fonction rénale comme paramètre principal de la gravité des maladies rénales. Méthodes de recherche rénale. Analyse clinique d'histoires de cas de patients atteints de maladies chroniques du système urinaire.
Signes de troubles et classification des maladies du système urinaire. Analyse clinique d'histoires de cas de patients atteints de maladies du système urinaire et leur analyse. L’importance des tests de la fonction rénale pour poser le bon diagnostic.
travail de cours, ajouté le 14/04/2016
L'importance du système excréteur. La structure et les caractéristiques liées à l'âge du système urinaire des enfants d'âge préscolaire, le processus de formation de l'urine et l'acte de miction. Caractéristiques des maladies du système urinaire chez l'enfant et leur prévention.
test, ajouté le 06/09/2015
Caractéristiques morpho-fonctionnelles du système urinaire. Anatomie des reins. La structure des reins. Mécanisme de formation de l'urine. Apport sanguin aux reins. Dysfonctionnement du système urinaire dû à une pathologie, pyélonéphrite. Méthodes d'examen de la fonction urinaire et rénale.
résumé, ajouté le 31/10/2008
Troubles du développement des organes du système génito-urinaire, qui ne permettent pas au corps de fonctionner normalement. Difficulté d'écoulement de l'urine, ses causes et ses conséquences. Agénésie rénale, ses types, méthodes de diagnostic et de traitement. Concept et causes de la dystopie rénale.
résumé, ajouté le 16/09/2014
Les tâches principales d'une infirmière sont d'organiser et de prodiguer des soins infirmiers. Mise en place d'un plan de soins pour les patients atteints de maladies des voies urinaires. Structure du système urinaire. Remplir et examiner le dossier infirmier du patient.
ORGANES URINAIRES. EMBRYOPHÉTOGENÈSE. ANOMALIES DE DÉVELOPPEMENT.
DÉVELOPPEMENT DU SYSTÈME URINAIRE
L'embryophétogenèse du système urinaire humain est en plusieurs étapes et extrêmement complexe. Les voies urinaires supérieures sont un dérivé du mésoderme (troisième couche germinale)
Stades de développement des reins :
1 – pronéphros – préférence
2 – mésonéphros – rein primaire
3 – métanéphros – secondaire(constante finale) bourgeon
Le pronéphros et le mésonéphros, bien qu'ils se développent dès les premiers stades de l'embryogenèse, ne sont qu'une récapitulation (répétition) des étapes de la phylogenèse humaine. Aux stades ultérieurs, soit ils sont complètement réduits (pré-rein), soit, après un fonctionnement à court terme, ils servent de canaux excréteurs des organes génitaux masculins (rein primaire).
Tous les types de reins se développent à partir de néphrotomes(néphragonadotomes, tissu métanéphrogénique) – pattes segmentaires se connectant somites– segments du mésoderme dorsal – avec splanchnotome– partie ventrale non segmentée du mésoderme. Splanchnotome comporte deux plaques latérales : somatopleure– la plaque latérale extérieure, et splanchnopleure(viscéropleura) - plaque latérale interne formant une cavité corporelle secondaire - en général(celom ; à partir de celui-ci se forment ensuite le péritoine, la plèvre, le péricarde). Chez l'embryon humain, la segmentation des néphrotomes n'est conservée que dans la partie crânienne.
Pronéphros – préférence(tête, rein antérieur) d'une personne existe de 3 à 6 semaines, fonctionne pendant environ 40 heures et se réduit rapidement. Tubules rénaux primitifs – protonéphridie, ouverture en total 2-3 en forme d'entonnoir néphrostomies, équipé d'un épithélium cilié (cils), manque de glomérules. Plusieurs artères afférentes partent de l'aorte, formant un glomérule vasculaire commun - glomérule pronéphrosus. Il est situé près de la paroi de la cavité du coelome, mais ne s'y ouvre pas. « L'urine » est filtrée dans son ensemble, où elle est « capturée » par l'épithélium cilié du néphrost, entrant dans le canal excréteur pronéphrostique le long de la protonéphridie. Les conduits situés caudalement fusionnent pour former le canal de Wolff (canal de Leiden-Leyden), qui se jette dans le canal primaire. cloaque.
Mésonéphros– rein primaire, corps de Wolff, corps d’Oken. Il se développe du néphrotome caudal au pronéphros et fonctionne pendant 2,5 à 3 mois. 20-30 méta-néphridium Le mésonéphros est plus long et plus alambiqué que la protonéphridie. L'extrémité proximale des métanéphridies se termine aveuglément, formant la capsule du mésonéphros, à partir de laquelle elle commence canaux mésonéphros- baignoire. mésonéphrose. Les canaux mésonéphros débouchent sur canal méso-néphrique– conduit. mésonéphricus ( Canal de Wolff). De l'aorte à la capsule du mésonéphros en dehors du coelome ils s'approchent amener des navires– les vaisseaux afférents, inclus dans la capsule de mésonéphridie et formant club vasculaire lunettes – glomérule mésonéphrosus. Le glomérule mésonéphros constitue avec la capsule corpuscule rénal(corpuscula renalis). L'ensemble des corpuscules et des conduits constitue Corps de loup- il filtre l'urine, qui pénètre ensuite dans le canal mésonéphrique (de Wolff) et, plus loin, dans l'allantoïde - le rudiment de la vessie. A la fin du 3ème mois, les tubules mésonéphros sont réduits.
Métanéphros– rein secondaire (définitif, permanent, pelvien). Se développe à partir de deux rudiments : blastème métanéphrogène(néphrotomes caudaux non segmentés) et extrémité crânienne excroissance urétérale(Kupffer - canal), qui est formé à partir de la partie inférieure du canal mésonéphrique (Wolffien). De là se forment le bassinet rénal, les grands et petits calices rénaux et les canaux collecteurs. La croissance de l'excroissance urétérale dans un tissu indifférencié induit la différenciation du blastème métanéphrogène : le néphron, y compris les corpuscules rénaux et les tubules néphroniques, se développe à partir du tissu métanéphrogénique (le processus est long ; au moment de la naissance, il existe jusqu'à 15 générations de néphrons). . Presque immédiatement après la formation, le rein secondaire s'élève et, au bout de 3 mois, se situe au-dessus du corps de Wolff, qui s'est alors considérablement atrophié. « En grandissant » dans l’amas de cellules mésenchymateuses (future graisse « brune ») de l’espace rétropéritonéal, le rein induit la formation de sa propre capsule graisseuse – para-néphrie, et le fascia rénal, le délimitant des espaces cellulaires de la région rétropéritonéale. Le sous-développement (ou retard de développement) du rein et les aberrations du processus de migration sont la principale cause d'anomalies de la position du rein et de son appareil de fixation.
La migration du rein s'accompagne de sa rotation progressive autour d'un axe vertical de sorte que le bassin, initialement situé ventro-latéralement, se révèle médial au parenchyme rénal. La fin de la migration s'accompagne de la divergence des pôles inférieurs des reins. L'artère rénale extra-organique est formée par la fusion et la réduction partielle de plusieurs troncs artériels, y compris mésonéphriques. Ce processus est associé (synchrone) à la migration des reins.
Le rein secondaire commence à fonctionner dans la seconde moitié de la vie embryonnaire.
Morphogenèse embryofœtale des malformations des reins et des uretères (Ayvazyan A.V., Voino-Yasenetsky A.M., 1988)
Développement de la vessie
La formation de la vessie humaine, comme celle de tous les mammifères supérieurs, se produit à partir de plusieurs parties se développant séquentiellement : partie ventrale du cloaque primaire et du sac urinaire(allantoïde) avec canal urinaire(urachus'a).
Au cours de la 2ème semaine de développement intra-utérin: à partir de la partie caudale de l'intestin postérieur primaire, se développant ventralement, se forme l'allantoïde (allantoïde) - « sac urinaire ».
A la 3ème semaine de développement intra-utérin: grandissant dans le sens caudal et s'étendant, intestin postérieur primaire formes cloaque. Dans celui-ci, entre l'allantoïde et l'intestin primaire, le pli urorectal. Dans le même temps, l'allantoïde, s'allongeant dans le sens proximal, forme canal urinaire(urachus), se dirigeant vers le nombril. Petit à petit, le sillon urogénital se rapproche membrane cloacale, divisant le cloaque en deux parties : ventrale - sinus génito-urinaire et dorsale - rectum. Entre le sinus urogénital et le rectum de pli rectal urinaire un septum situé frontalement se forme (futur Façade Salishchev-Denonvilliers). La couche viscérale du péritoine, passant de l'allantoïde (future vessie) au rectum, forme évidement recto-vésical– excavation rectovésicale. Au fil du temps, la membrane cloacale disparaît et deux ouvertures se forment : ouverture du sinus urogénital– l’ostium urogénitale, menant au sinus urogénital – la partie ventrale du cloaque, et anus– l'anus, menant au rectum – la partie dorsale du cloaque. Les canaux mésonéphriques (ductus mesonephricus) se transforment en allantoïde, à partir duquel se développent les uretères et les canaux déférents.
Canal urinaire– ouraque – à la fin de la vie intra-utérine, il est partiellement oblitéré et se transforme en ligament ombilical médian. Si cela ne se produit pas, divers défauts se forment, nécessitant généralement une intervention chirurgicale :
Fistule ombilicale – non-fermeture du canal urinaire proximal ;
Fistule vésico-ombilicale – non-fermeture de tout le canal urinaire ;
Kyste des voies urinaires - les voies urinaires ne sont pas fermées dans le tiers moyen,
Le diverticule vésical est une non-fermeture du canal urinaire distal.
Des perturbations importantes dans les processus de différenciation et de développement des sections ventrales de l'extrémité caudale de l'embryon au stade de la formation de l'allantoïde et de la division du cloaque primaire conduisent à des défauts anatomiques grossiers - exstrophie(1:40 000 nouveau-nés ; garçons/filles – 2/1) vessie (toujours accompagnée de l'absence de symphyse), formation de fistules rectovésicales (ou rectovaginales), etc.
1. 7. ANOMALIES DE DÉVELOPPEMENT DES REINS ET DES VOIES URINAIRES SUPÉRIEURES
L'embryophétogenèse complexe des organes urinaires humains est une raison indirecte du fait qu'environ 40 % de tous les défauts de développement décrits chez l'homme sont des dérivés du sinus urogénital. Le fait est évident : plus l'impact tératogène s'est produit tôt, moins le génome de l'organisme en développement est adaptatif, plus l'anomalie de développement est grave.
Les anomalies du système urinaire sont réparties dans les groupes suivants : anomalies du nombre de reins : agénésie bilatérale et unilatérale, double rein ; anomalies des reins : dystopie homolatérale (prolapsus du rein), dystopie hétérolatérale (croisée) (prolapsus du rein et son déplacement vers le côté opposé du corps) ; anomalies dans la position relative des reins : reins en forme de fer à cheval, en biscuit, en S, en L ; anomalies de la taille et de la structure des reins : aplasie, hypoplasie, rein polykystique ; anomalies du bassinet du rein et des uretères : kystes, diverticules, bifurcation du bassin, anomalies du nombre, du calibre, de la forme et de la position des uretères. Beaucoup de ces anomalies provoquent le développement de calculs rénaux, d'inflammations (pyélonéphrite), d'hypertension artérielle. L'influence de diverses formes d'anomalies du système urinaire sur le corps d'un enfant peut s'exprimer de différentes manières. Si certains troubles entraînent le plus souvent la mort intra-utérine d'un bébé ou sa mort en bas âge, alors un certain nombre d'anomalies n'ont pas d'effet significatif sur le fonctionnement de l'organisme, et ne sont souvent découvertes que par hasard lors d'examens médicaux de routine. une anomalie qui ne dérange pas l'enfant peut provoquer de graves troubles fonctionnels à l'âge adulte, voire à la vieillesse.
Comment prévenir les anomalies du système urinaire ? On pense que le risque de développer de tels troubles est plus élevé au cours des premiers mois de la grossesse, lorsque les principaux organes, y compris le système urinaire, se forment. La future maman ne doit prendre aucun médicament sans prescription médicale. En cas de rhume et d'autres maladies entraînant une forte fièvre et une intoxication, il est conseillé de se rendre immédiatement à l'hôpital lors de la planification d'une grossesse. Il est conseillé aux jeunes parents de se soumettre à un examen médical qui permettra d'exclure diverses maladies entraînant des anomalies. fœtus. S'il y a déjà eu des cas d'anomalies dans la famille, une consultation avec un généticien est nécessaire.
Lors de la formation du fœtus dans l'utérus, divers dysfonctionnements peuvent survenir, qui se manifestent par la présence de diverses anomalies du système génito-urinaire. Dans une telle situation, un ou plusieurs organes peuvent être endommagés en même temps.
De tels écarts peuvent être détectés par hasard, car ils ne posent aucun problème ni n'entraînent la mort du bébé avant la naissance. Elles représentent environ 40 % de toutes les anomalies intra-utérines.
Pathologies de la vessie
Parmi les maladies congénitales de l'organe, on distingue les anomalies :
Certaines anomalies peuvent ne jamais se manifester
- Diverticule. Caractérisé par une saillie du mur vers l'extérieur. Dans ce cas, il se produit une accumulation et une stagnation de l'urine, ce qui entraîne divers types d'inflammation et la formation de calculs. Dans certains cas, les diverticules sont plus gros que l’organe lui-même. Ils peuvent être congénitaux ou acquis. Ils sont classés en simples et multiples, vrais et faux. Avec ce dernier type, on observe une formation herniaire ronde de la membrane muqueuse.
- Anomalie ourachale. Sa formation s'arrête pendant la période prénatale, ce qui affecte le processus de sa fusion. Cela peut provoquer la formation d’une fistule d’où seront libérés de l’urine ou du pus, ainsi que des kystes.
- . L'une des déviations dangereuses est que son apparition entraîne la mort en raison de l'absence d'organe.
- Exstrophie. Il n'y a pas de parois antérieures de la vessie, de l'urètre ou de la cavité abdominale, donc dans la partie inférieure de l'abdomen se trouve un trou d'un diamètre égal à plusieurs centimètres. Il montre l'arrière de la vessie et l'embouchure de l'uretère, par lequel l'urine est évacuée. Cela provoque l’absence de formation cartilagineuse du squelette et le placement des hanches médialement. Cela provoque l’écoulement de l’urine, provoquant le développement d’ulcères et d’inflammations. Lorsque la pathologie survient chez le premier enfant, elle se manifeste également chez le second.
- Doubler. Dans ce cas, la vessie est divisée par un septum en 2 moitiés, chacune possédant son propre uretère. La pathologie est diagnostiquée assez rarement. Une distinction est faite entre le doublement complet et incomplet. Dans le premier cas, l'urètre et le col de la vessie sont doublés, ce qui dans 50 % des cas s'accompagne d'une fistule vaginale et d'un double rectum. Le deuxième type de pathologie est caractérisé par une vessie à deux chambres.
- Contracture cervicale. La pathologie est également rare et se manifeste par une prolifération accrue de tissus fibreux dans les couches sous-muqueuses et musculaires.
Anomalies du système génito-urinaire
Les pathologies des uretères sont enregistrées chez un cinquième de la population et ne sont pas détectées immédiatement, mais grâce à des mesures diagnostiques :
- Doubler. Avec le type complet, ils se croisent et s'ouvrent avec leur bouche dans la vessie, et avec le type incomplet, ils fusionnent.
- Uretérocèle. Accompagné d'une saillie de la partie la plus distale de l'uretère dans la vessie.
- Dysplasie neuromusculaire. Elle se caractérise par un sous-développement du système neuromusculaire et une région intra-muros étroite.
- Uretère rétrocave. La localisation de la partie médiane de l'uretère (principalement la droite) derrière la veine cave inférieure est diagnostiquée.
- L'ostium ectopique implique sa localisation inhabituelle.
- Uretère spiralé.
Les anomalies suivantes dans le développement de l'urètre :
- Les valvules congénitales sont des formations pliées simples ou multiples sous la forme de cavaliers à l'intérieur de l'organe.
- Hypertrophie du tubercule séminal. Une anomalie rare pouvant entraîner une fermeture complète de la lumière de l'urètre.
- Rétrécissement congénital.
- Hypospadias. Il n'y a pas de mur arrière du canal ou son absence partielle. Selon la localisation de l'ouverture d'excrétion, on distingue les pathologies telles que scrotales, tiges, périnéales et capitées (50 % des cas).
- Épispadias. Il s'agit d'une non-fermeture de la paroi antérieure de l'urètre. Elle peut être totale, tige et capitée (garçons), clitoridienne (filles).
Les écarts dans la formation des reins sont classés en fonction du nombre, de la taille, de l'emplacement, de la structure et de la relation avec les organes. Diagnostiqué chez 30% des patients atteints de maladies urologiques.

Au troisième mois de grossesse, des anomalies du développement fœtal peuvent être détectées
On distingue les types de troubles suivants :
- L'agénésie est l'absence totale de reins. Combiné avec des anomalies des organes génitaux masculins.
- Aplasie. Absence du bassin, qui l'empêche de produire de l'urine. Si les pathologies sont bilatérales, elles entraînent une mort fœtale.
- Doubler. La forme complète de l'anomalie implique la présence de systèmes pyélocalicaux dans chaque partie du rein, dans la partie supérieure il est sous-développé et d'uretères. Le doublement des vaisseaux et du parenchyme du rein est une forme incomplète.
- Présence d'un troisième rein. Sa taille est plus petite que celle habituelle, mais cela n'affecte pas le fonctionnement.
- Hypoplasie. L’artère rénale étant petite, elle produit moins d’urine.
- La dystopie est une position incorrecte des reins les uns par rapport aux autres ou au squelette.
- Fusion des reins par les pôles supérieur, inférieur (en fer à cheval) ou opposés (en L ou en S). Une forme semblable à un biscuit est observée lorsque la fusion se produit le long de la nervure interne.
- Avec la dysplasie, on observe une diminution de la taille de l'organe et une structure non standard du parenchyme (rudimentaire et nain).
- Multikystique. Le parenchyme est remplacé par des kystes, il n'y a pas d'artère rénale, l'uretère est fermé à une extrémité.
- Bourgeon spongieux. La pathologie bilatérale est associée à la formation de kystes.
Parmi les anomalies associées à des modifications de la structure des reins, on distingue les kystes polykystiques, les kystes solitaires et dermoïdes et le diverticule du bassinet du rein. Le premier est le plus courant et est à l’origine du développement de l’insuffisance rénale.
Les médecins classent également séparément les pathologies de formation des vaisseaux rénaux. Ils diffèrent par le nombre, la forme et l’emplacement de ces veines et artères et peuvent provoquer le développement de maladies dangereuses.
Déviations dans la formation du système reproducteur
Pour les garçons:
- rétrécissement du prépuce (phimosis);
- bride courte;
- localisation anormale du testicule (cryptorchidie et ectopie) ;
- anorchisme (absence de deux testicules), monorchisme (1 testicule) et polyorchisme (3 testicules ou plus) ;
- hypoplasie testiculaire – dimensions inférieures à 1 cm.

Grâce aux méthodes de diagnostic, vous pouvez détecter le problème à temps
Pour les filles:
- structure anormale de la cloison vaginale;
- infantilisme - développement retardé des organes génitaux;
- double utérus ou vagin;
- l'hymen n'a pas d'ouverture ;
- structure anatomique anormale de l'utérus (à une ou deux cornes, en forme de selle).
Ces maladies provoquent l’infertilité et les femmes deviennent incapables de porter un fœtus.
Symptômes d'anomalies rénales
Principaux signes de pathologies :
- miction douloureuse, incontinence urinaire;
- douleur à gauche (droite) ou des deux côtés, région pelvienne, avec dystopie thoracique, elles surviennent dans l'abdomen après avoir mangé ;
- le fonctionnement des intestins et de l'estomac, l'apport sanguin aux organes est perturbé;
- saignement;
- augmentation de la pression artérielle et de la température corporelle;
- rhumes fréquents;
- nausée et vomissements;
- gonflement des membres inférieurs;
- urine trouble mélangée à du sang ou du pus ;
- taux élevés de créatinine et d'urée dans le sang.
De nombreux symptômes apparaissent dus à l'ajout d'infections et au développement de pathologies rénales telles que la pyélonéphrite, la lithiase urinaire.
Causes des anomalies
Les médecins ont identifié un certain nombre de facteurs influençant le développement pathologique du système génito-urinaire chez le fœtus :

Types d'anomalies rénales
- hérédité;
- maladies infectieuses ou vénériennes chez la mère pendant la grossesse ;
- consommation excessive de drogues, d'alcool et de nicotine ;
- exposition aux radiations ou empoisonnement par des poisons toxiques ;
- abus de médicaments hormonaux;
- activité professionnelle nuisible.
Diagnostique
Toutes les anomalies ne sont pas visibles à la naissance d'un enfant ; si des symptômes apparaissent ou si des anomalies dans la formation de parties du système génito-urinaire sont suspectées, il est nécessaire de subir une série d'examens :

L'opération peut résoudre le problème. C'est le moyen le plus sûr de traiter les pathologies
- Radiographie avec introduction d'un agent de contraste ;
- Cytoscopie et urétroscopie ;
- urographie excrétrice;
- artériographie rénale sélective ;
- angiographie;
- pyélographie ou cystographie rétrograde ;
- Échographie des organes pelviens ;
- TDM ou IRM ;
- scintigraphie;
- fistulographie.
Le médecin peut déterminer certaines anomalies après avoir examiné l'enfant, mais la plupart sont détectées lors d'examens visant à détecter la présence de maladies du système génito-urinaire.
Traitement des anomalies du système génito-urinaire
Étant donné que la plupart des pathologies affectent le débit urinaire normal, la principale méthode de traitement est la chirurgie. Certaines opérations ne sont pratiquées qu'à partir d'un certain âge, d'autres sont prescrites lorsque le traitement médicamenteux s'avère inefficace. Un tel traitement n'apporte pas toujours une solution à 100 % au problème ; des interventions chirurgicales répétées peuvent parfois être nécessaires. Pour les anomalies des organes génitaux, des médicaments hormonaux sont prescrits.
Pendant la période postopératoire, le patient doit suivre toutes les prescriptions du médecin, se présenter à temps aux examens et passer des tests pour vérifier la fonction rénale.
Les anomalies dans le développement des organes entraînent la mort du fœtus ; si la pathologie identifiée n'est pas mortelle, elle nécessite alors un traitement rapide.