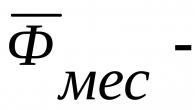Vedecký prístup
Objektívne povedané, žiadny rodič nie je voči tomuto riziku imúnny. Každý z nás nesie v priemere 10-12 defektných génov, ktoré sme dostali od našich príbuzných a možno ich prenesieme na vlastné deti. Dnes veda pozná asi 5 000 dedičných chorôb, ktoré vznikajú v dôsledku problémov v ľudskom genetickom aparáte – v génoch alebo chromozómoch.
Delia sa do troch hlavných skupín: monogénne, polygénne a chromozomálne.
Dnes je možné z genetického hľadiska vysvetliť takmer akúkoľvek patológiu. Chronická tonzilitída je dedičná porucha imunity, cholelitiáza je dedičná metabolická porucha.
Druhy chorôb
Monogénne ochorenia sú spôsobené defektom jedného génu. Dnes je známych asi 1400 takýchto ochorení. Ich prevalencia je síce nízka (5 – 10 % z celkového počtu dedičných ochorení), ale úplne nevymiznú. Medzi najčastejšie v Rusku patrí cystická fibróza, fenylketonúria, adrenogenitálny syndróm a galaktozémia. Na identifikáciu uvedených patológií sa všetci novorodenci u nás podrobujú špeciálnym testom (žiaľ, žiadna krajina na svete nemôže testovať deti na prítomnosť všetkých defektných génov). Ak sa zistí odchýlka, dieťa sa prenesie na špeciálnu diétu, ktorú je potrebné dodržiavať do 12 rokov, niekedy až do 18 rokov. Ak sa chorým rodičom narodia zdravé deti, potom budú všetci ich potomkovia „bez chyby“.
Polygénne (alebo multifaktoriálne) ochorenia sú spojené s narušením interakcie viacerých génov, ako aj faktorov prostredia. Ide o najväčšiu skupinu – zahŕňa asi 90 % všetkých dedičných ľudských chorôb, ich prevenciu a následnú liečbu.
Prenosové cesty
Hlavným prenášačom choroby je chorá matka alebo otec. Ak chorobou trpia obaja, riziko sa niekoľkonásobne zvyšuje. Zároveň, aj keď ste vy a váš manželský partner zdraví, vo vašom tele je množstvo defektných génov. Sú jednoducho potlačení tými normálnymi a sú „tichí“. Ak máte vy a váš manžel rovnaký „tichý“ gén, u vašich detí sa môže vyvinúť dedičná choroba.
Choroby, ktoré sú „spojené s pohlavím“, majú svoju vlastnú zvláštnosť dedičnosti - hemofília, Guntherova choroba. Sú riadené génmi, ktoré sa nachádzajú na pohlavnom chromozóme. Rodičia pacienta majú určité typy onkológie, vývojové chyby (vrátane rázštepu pery a podnebia). V niektorých prípadoch rodičia prenášajú nie samotnú chorobu, ale predispozíciu k nej (diabetes mellitus, ischemická choroba srdca, alkoholizmus). Deti dostávajú nepriaznivú kombináciu génov, ktorá za určitých podmienok (stres, vážny úraz, zlé životné prostredie) môže viesť k rozvoju ochorenia. Navyše, čím výraznejšia je choroba u mamy alebo otca, tým vyššie je riziko.
Obrovské množstvo času a úsilia sa venuje ľudským chromozomálnym dedičným ochoreniam, ich prevencii a liečbe vznikajú v dôsledku zmien v počte a štruktúre chromozómov. Napríklad najznámejšia anomália – Downova choroba – je dôsledkom strojnásobenia 2. chromozómu. Takéto mutácie nie sú také zriedkavé, vyskytujú sa u 6 novorodencov. Ďalšími bežnými chorobami sú Turnerov, Edwardsov a Patauov syndróm. Všetky sú charakterizované viacerými poruchami: oneskorený fyzický vývoj, mentálna retardácia, poruchy kardiovaskulárneho, urogenitálneho, nervového a iného systému. Liek na chromozomálne abnormality zatiaľ nebol nájdený.
dieťa môže byť zdravé, ale ak je matka nositeľkou mutantného génu, pravdepodobnosť, že sa narodí chorý chlapec, je 5°%. Dievčatá sa rodia zdravé, no polovica z nich sa zasa stane nositeľkami defektného génu. Chorý otec neprenáša chorobu na svojich synov. Dcéry môžu ochorieť len vtedy, ak je matka prenášačka.
Z egyptskej hrobky
Starovekí zobrazovali faraóna Achnatona a kráľovnú Nefertiti s dosť nezvyčajným vzhľadom. Ukazuje sa, že nejde len o umelecké videnie maliarov. Na základe neprirodzene pretiahnutého „vežovitého“ tvaru lebky, malých očí, abnormálne dlhých končatín (takzvané „pavúčie prsty“) a nedefinovanej brady („vtáčia tvár“) vedci identifikovali Minkowského-Shafarov syndróm, z dedičných typov anémie (anémia).
Z ruských dejín
Dedičný charakter má aj porucha zrážanlivosti krvi (hemofília) u syna posledného ruského cára Mikuláša II., careviča Alexeja. Toto ochorenie sa prenáša cez materskú líniu, ale vyskytuje sa výlučne u chlapcov. S najväčšou pravdepodobnosťou bola prvou majiteľkou génu pre hemofíliu kráľovná Viktória Veľkej Británie, Alexejova prababička.
Ťažkosti pri detekcii
Nie vždy sa dedičné choroby objavujú od narodenia. Niektoré typy mentálnej retardácie sa prejavia až vtedy, keď dieťa začne rozprávať alebo začne chodiť do školy. Ale Gettingtonovu choreu (druh progresívnej mentálnej retardácie) možno vo všeobecnosti rozpoznať až po rokoch.
Okrem toho sa „tiché“ gény môžu stať nástrahami. Ich účinok sa môže prejaviť počas celého života – vplyvom negatívnych vonkajších faktorov (nezdravý životný štýl, užívanie množstva liekov, radiácia, znečistenie životného prostredia). Ak je vaše bábätko ohrozené, môžete absolvovať molekulárne genetické vyšetrenie, ktoré pomôže identifikovať pravdepodobnosť vzniku ochorenia v každom konkrétnom prípade. Ďalej môže odborník predpísať preventívne opatrenia. Ak sa ukáže, že choré gény sú dominantné, je nemožné vyhnúť sa chorobe. Môžete len zmierniť príznaky ochorenia. Ešte lepšie je pokúsiť sa ich varovať pred momentom narodenia.
Riziková skupina
Ak máte vy a váš manžel jeden z uvedených faktorov, je lepšie absolvovať genetické poradenstvo ešte pred tehotenstvom.
1. Prítomnosť niekoľkých prípadov dedičných chorôb na oboch líniách. Aj keď ste zdraví, môžete byť nositeľmi defektných génov.
2. Vek nad 35 rokov. V priebehu rokov sa počet mutácií v tele hromadí. Riziko mnohých chorôb rastie exponenciálne. Takže pri Downovej chorobe pre 16-ročné matky je to 1:1640, pre 30-ročné - 1:720, pre 40-ročné - už 1:70.
3. Narodenie predchádzajúcich detí so závažnými dedičnými chorobami.
4. Niekoľko prípadov potratu. Často sú spôsobené vážnymi genetickými alebo chromozomálnymi abnormalitami u plodu.
5. Dlhodobé užívanie liekov ženou (antikonvulzíva, lieky proti štítnej žľaze, protinádorové lieky, kortikosteroidy).
6. Kontakt s toxickými a rádioaktívnymi látkami, ako aj alkoholizmus a drogová závislosť. To všetko môže spôsobiť genetické mutácie.
Vďaka pokroku v medicíne majú teraz všetci rodičia na výber, či v rodinnej anamnéze vážneho ochorenia pokračovať, alebo ho ukončiť.
Metódy prevencie
Ak ste ohrození, mali by ste sa poradiť s genetikom. Na základe podrobného rodokmeňa a ďalších údajov rozhodne, či sú vaše obavy oprávnené. Ak váš lekár potvrdí, že ste v ohrození, mali by ste podstúpiť genetické vyšetrenie. Zistí, či ste nositeľmi nebezpečných defektov.
Ak je riziko chorého dieťaťa príliš vysoké, odborníci odporúčajú namiesto prirodzeného počatia uchýliť sa k in vitro fertilizácii (IVF) s preimplantačnou genetickou diagnostikou (PGD). PGD umožňuje jednej bunke odobranej z embrya určiť, či je zdravé alebo choré. Potom sa vyberú a implantujú do maternice iba zdravé embryá. Po IVF je miera tehotenstva 40% (môže byť potrebný viac ako jeden postup). Malo by sa pamätať na to, že embryo je testované na konkrétnu chorobu (pre ktorú je vopred identifikované zvýšené riziko). To neznamená, že výsledné dieťa je zaručené proti iným chorobám, vrátane dedičných. PGD je zložitá a drahá metóda, ale v správnych rukách funguje dobre.
Počas tehotenstva by ste mali určite navštevovať všetky rutinné ultrazvuky a darovať krv na „trojitý test“ (na posúdenie stupňa rizika rozvoja patológie). Ak existuje riziko chromozomálnych mutácií, môžete podstúpiť biopsiu choriových klkov. Aj keď existuje riziko ukončenia tehotenstva, táto manipulácia umožňuje určiť prítomnosť chromozomálnych abnormalít. Ak sa zistia, odporúča sa prerušiť tehotenstvo.
Dedičné choroby sú choroby spôsobené chromozomálnymi a génovými mutáciami. Veda, ktorá študuje fenomény dedičnosti a variability v ľudských populáciách, je genetika. Často sa verí, že pojmy „dedičná choroba“ a „vrodená choroba“ sú synonymá. Avšak na rozdiel od vrodených chorôb, ktoré sa vyskytujú pri narodení dieťaťa, dedičné choroby sú už spôsobené dedičnými a exogénnymi faktormi.
Problémy dedičnosti zaujímali ľudí už mnoho storočí. Napríklad taká choroba ako hemofília je známa už dlho. V tomto ohľade boli manželstvá medzi pokrvnými príbuznými zakázané. Mnohí vedci predložili svoje hypotézy o výskyte dedičných patológií. Ich predpoklady neboli vždy založené na vedeckých pozorovaniach. Až v 20. storočí s rozvojom genetiky boli odhalené vedecké dôkazy.
Pokrok v oblasti medicíny viedol k relatívnemu zvýšeniu podielu geneticky podmienených patológií. Doteraz bolo identifikovaných viac ako 3500 dedičných ľudských chorôb. Asi 5 % detí sa rodí s genetickými alebo vrodenými chorobami.
Z genetického hľadiska možno všetky choroby s dedičnými a environmentálnymi faktormi vo vývoji rozdeliť do 3 skupín:
- Dedičné ochorenia s fenotypovou mutáciou, ktoré sú takmer nezávislé od prostredia. Ide spravidla o genetické a chromozomálne dedičné ochorenia, ako je hemofília, Downova choroba, fenylketonúria a iné.
- Choroby s dedičnou predispozíciou, ktorých prejav si vyžaduje vplyv vonkajšieho prostredia. Medzi takéto ochorenia patrí diabetes mellitus, dna, ateroskleróza, peptický vred, psoriáza, hypertenzia atď.
- Choroby, pri vzniku ktorých dedičnosť nehrá rolu. Patria sem zranenia, popáleniny a akékoľvek infekčné choroby.
Choroby spôsobené zmenami v štruktúre chromozómov sa nazývajú chromozomálne choroby. Ochorenia spôsobené zmenami v štruktúre DNA sa nazývajú génové choroby. Klinická diagnostika dedičných chorôb je založená na klinickom, genealogickom a paraklinickom vyšetrení.
Treba si uvedomiť, že donedávna boli takmer všetky dedičné choroby považované za nevyliečiteľné. Dnes sa však všetko zmenilo. Diagnostikovaním chorôb v skorých štádiách dokážem ľuďom zmierniť utrpenie a niekedy ich dokonca zbaviť. Vďaka genetike dnes existuje veľa expresných diagnostických metód, napríklad biochemické testy a imunologické metódy. Jasným príkladom je schopnosť modernej medicíny bojovať proti detskej obrne.
Zoznam genetických chorôb * Hlavné články: dedičné choroby, Dedičné metabolické choroby, Enzymopatia. * Vo väčšine prípadov je uvedený aj kód označujúci typ mutácie a s ňou spojené chromozómy. tiež... ... Wikipedia
Nižšie je uvedený zoznam symbolických stužiek (symbolická alebo oznamovacia stuha, z anglického Awareness ribbon) malý kúsok pásky zložený do slučky; používa sa na demonštráciu postoja nosiča pásky k akémukoľvek problému alebo... ... Wikipedia
Táto stránka je glosár. Pozri tiež: Zoznam genetických malformácií a chorôb Genetické pojmy v abecednom poradí... Wikipedia
Servisný zoznam článkov vytvorených na koordináciu práce na vývoji témy. Toto varovanie nie je nastavené... Wikipedia
Odvetvie humánnej genetiky venované štúdiu úlohy dedičných faktorov v ľudskej patológii na všetkých hlavných úrovniach organizácie života od populácie až po molekulárnu genetiku. Hlavná sekcia M.g. pozostáva z klinickej genetiky,...... Lekárska encyklopédia
Dedičné choroby sú choroby, ktorých výskyt a vývoj je spojený s poruchami programovacieho aparátu buniek, ktoré sa dedia prostredníctvom gamét. Termín sa používa vo vzťahu k polyetiologickým ochoreniam, na rozdiel od ... Wikipedia
Choroby, ktorých výskyt a vývoj sú spojené s poruchami programovacieho aparátu buniek, ktoré sa dedia prostredníctvom gamét. Termín sa používa v súvislosti s polyetiologickými ochoreniami, na rozdiel od užšej skupiny Genetické... ... Wikipedia
Dedičné ochorenie je ochorenie, ktorého výskyt a vývoj je spojený s poruchami programovacieho aparátu buniek, ktoré sa dedia prostredníctvom gamét. Termín sa používa v súvislosti s polyetiologickými ochoreniami, na rozdiel od ... ... Wikipédie
Dedičné metabolické poruchy zahŕňajú veľkú skupinu dedičných ochorení ovplyvňujúcich metabolické poruchy. Takéto poruchy tvoria významnú časť skupiny metabolických porúch (metabolických ochorení).... ... Wikipedia
knihy
- Detské choroby, Belopolsky Jurij Arkadevič. Zdravie dieťaťa v akomkoľvek veku je pre lekára osobitnou úlohou, pretože rastúci organizmus si vyžaduje väčšiu pozornosť a väčšiu ostražitosť vo vzťahu k chorobám. Plánované lekárske vyšetrenia, identifikácia...
- Úvod do molekulárnej diagnostiky a génovej terapie dedičných chorôb, V. N. Gorbunova, V. S. Baranov. Kniha načrtáva moderné predstavy o štruktúre ľudského genómu, metódach jeho štúdia, štúdiu génov, ktorých mutácie vedú k ťažkej dedičnej patológii: považované...
Dedičné choroby pediatri, neurológovia, endokrinológovia
A-Z A B C D E F G H I J J K L M N O P R S T U V X C CH W SCH E Y Z Všetky sekcie Dedičné choroby Pohotovostné stavy Choroby očí Detské choroby Mužské choroby Ženské choroby Kožné choroby Nervové choroby Reumatické choroby Urologické choroby Choroby endokrinné Imunitné choroby a lymfatické choroby nie Choroby chrupu Choroby krvi Choroby prsníkov Choroby a úrazy ODS Choroby dýchacích ciest Choroby tráviaceho systému Choroby srdca a ciev Choroby hrubého čreva Choroby ucha, hrdla, nosa Drogové problémy Duševné poruchy Poruchy reči Kozmetické problémy Estetické problémy
Dedičné choroby– veľká skupina ľudských chorôb spôsobených patologickými zmenami v genetickom aparáte. V súčasnosti je známych viac ako 6 tisíc syndrómov s dedičným mechanizmom prenosu a ich celková frekvencia v populácii sa pohybuje od 0,2 do 4 %. Niektoré genetické choroby majú špecifickú etnickú a geografickú prevalenciu, zatiaľ čo iné sa vyskytujú s rovnakou frekvenciou na celom svete. Štúdium dedičných chorôb je v prvom rade zodpovednosťou lekárskej genetiky, ale s takouto patológiou sa môže stretnúť takmer každý lekársky špecialista: pediatri, neurológovia, endokrinológovia, hematológovia, terapeuti atď.
Dedičné choroby by sa mali odlišovať od vrodených a rodinných patológií. Príčinou vrodených ochorení môže byť nielen genetika, ale aj nepriaznivé exogénne faktory ovplyvňujúce vyvíjajúci sa plod (chemické a liečivé zlúčeniny, ionizujúce žiarenie, vnútromaternicové infekcie a pod.). Zároveň sa nie všetky dedičné choroby prejavia hneď po narodení: napríklad príznaky Huntingtonovej chorey sa zvyčajne prvýkrát objavia vo veku nad 40 rokov. Rozdiel medzi dedičnou a rodinnou patológiou je v tom, že táto patológia nemusí byť spojená s genetickými, ale so sociálnymi, každodennými alebo profesionálnymi determinantmi.
Výskyt dedičných chorôb je spôsobený mutáciami - náhlymi zmenami v genetických vlastnostiach jedinca, čo vedie k objaveniu sa nových, nezvyčajných vlastností. Ak mutácie postihujú jednotlivé chromozómy, menia ich štruktúru (v dôsledku straty, získania, zmeny polohy jednotlivých úsekov) alebo ich počet, takéto ochorenia sa klasifikujú ako chromozomálne. Najčastejšími chromozomálnymi abnormalitami sú duodenálna a alergická patológia.
Dedičné ochorenia sa môžu objaviť hneď po narodení dieťaťa, ako aj v rôznych štádiách života. Niektoré z nich majú nepriaznivú prognózu a vedú k skorému úmrtiu, iné výrazne neovplyvňujú dĺžku a dokonca kvalitu života. Najťažšie formy dedičnej fetálnej patológie spôsobujú spontánny potrat alebo sú sprevádzané mŕtvo narodenými plodmi.
Vďaka pokroku v medicínskom vývoji možno dnes už pred narodením dieťaťa pomocou metód prenatálnej diagnostiky odhaliť asi tisíc dedičných chorôb. Posledne menované zahŕňajú ultrazvuk a biochemický skríning I (10-14 týždňov) a II (16-20 týždňov) trimestrov, ktoré sa vykonávajú všetkým tehotným ženám bez výnimky. Okrem toho, ak existujú ďalšie indikácie, môžu sa odporučiť invazívne postupy: biopsia choriových klkov, amniocentéza, kordocentéza. Ak sa spoľahlivo potvrdí skutočnosť ťažkej dedičnej patológie, žene sa ponúkne umelé prerušenie tehotenstva zo zdravotných dôvodov.
Všetci novorodenci v prvých dňoch života sú tiež podrobení vyšetreniu na dedičné a vrodené metabolické ochorenia (fenylketonúria, adrenogenitálny syndróm, vrodená adrenálna hyperplázia, galaktozémia, cystická fibróza). Pomocou cytogenetických, molekulárno-genetických a biochemických výskumných metód možno identifikovať iné dedičné choroby, ktoré neboli rozpoznané pred alebo bezprostredne po narodení dieťaťa.
Úplné vyliečenie dedičných chorôb v súčasnosti bohužiaľ nie je možné. Medzitým pri niektorých formách genetickej patológie možno dosiahnuť významné predĺženie života a zabezpečenie jeho prijateľnej kvality. Pri liečbe dedičných ochorení sa používa patogenetická a symptomatická terapia. Patogenetický prístup k liečbe zahŕňa substitučnú liečbu (napríklad krvnými koagulačnými faktormi pri hemofílii), obmedzenie používania niektorých substrátov pri fenylketonúrii, galaktozémii, chorobe javorového sirupu, doplnenie deficitu chýbajúceho enzýmu alebo hormónu a pod. použitie širokej škály liekov, fyzioterapie, rehabilitačných kurzov (masáže, cvičebná terapia). Mnohí pacienti s genetickou patológiou už od raného detstva potrebujú korekčné a vývojové kurzy u logopéda a logopéda.
Možnosti chirurgickej liečby dedičných ochorení sa redukujú najmä na odstraňovanie ťažkých malformácií, ktoré zasahujú do normálneho fungovania organizmu (napr. korekcia vrodených srdcových chýb, rázštepov pery a podnebia, hypospádie a pod.). Génová terapia dedičných chorôb má stále skôr experimentálny charakter a má ešte ďaleko od širokého využitia v praktickej medicíne.
Hlavným smerom prevencie dedičných chorôb je lekárske genetické poradenstvo. Skúsení genetici sa poradia s manželským párom, predpovedajú riziko vzniku potomkov s dedičnou patológiou a poskytnú odbornú pomoc pri rozhodovaní o splodení dieťaťa.
9.1 Pojem, klasifikácia a znaky dedičnej patológie
Patológia je akákoľvek odchýlka od normálneho priebehu biologických procesov - metabolizmus, rast, vývoj, reprodukcia.
Dedičná patológia je odchýlka od normy so stanoveným faktom dedičnosti, to znamená prenos z generácie na generáciu. Je potrebné rozlišovať medzi vrodenou patológiou – prítomnou od narodenia jedinca – a dedičnou patológiou. Vrodená patológia môže byť spôsobená pôsobením faktorov prostredia – nedostatok živín a kyslíka počas vnútromaternicového vývoja, pôrodné poranenia, infekcie a pod. Stanovenie skutočnosti dedičnosti abnormálnej vlastnosti v súlade s požiadavkami genetickej analýzy (kapitola II) je jediným základom na rozpoznanie dedičnej povahy patológie.
Existujú dva typy klasifikácie dedičnej patológie. Prvým (akceptovaným hlavne v domácej literatúre) je klinický typ. Podľa tohto typu klasifikácie existujú štyri skupiny chorôb:
Skupina I sú vlastne dedičné choroby – chromozomálne a génové choroby (Edwardsov a Patauov syndróm, fenylketonúria, cystická fibróza);
Skupina II – ochorenia s výraznou dedičnou predispozíciou, v patogenéze ktorých je prejav dedičných faktorov determinovaný pôsobením špecifických vonkajších okolností (arteriálna hypertenzia, diabetes mellitus, dna);
Skupina III – choroby, ktoré sú determinované predovšetkým environmentálnymi faktormi, ale v patogenéze ktorých zohrávajú určitú úlohu dedičné faktory (glaukóm, ateroskleróza, rakovina prsníka);
Skupina IV – choroby, s ktorými dedičnosť na prvý pohľad nemá nič spoločné (otrava jedlom, zlomeniny, popáleniny).
Treba poznamenať, že často používané pojmy „familiárne“ a „sporadické“ ochorenia priamo nesúvisia s dedičnosťou. Rodinné choroby sa pozorujú u príbuzných, ale môžu byť spôsobené aj rovnakými vonkajšími príčinami, napríklad povahou výživy. Sporadické prípady sa vyskytujú u jednotlivých jedincov, ale môžu byť spôsobené aj zriedkavou kombináciou alel alebo mutáciou de novo.
Druhý klasifikačný systém – genetický – je v zahraničnej literatúre všeobecne akceptovaný a v poslednej dobe nachádza čoraz častejšie využitie v literatúre v ruštine. Podľa tohto systému existuje päť skupín:
Skupina I – génové ochorenia podmienené mutáciami určitých génov. Sú to prevažne monogénne znaky s autozomálne dominantným, autozomálne recesívnym, pohlavne viazaným dominantným, pohlavne viazaným recesívnym, holandrickým a mitochondriálnym spôsobom dedičnosti (kapitola II);
Skupina II – chromozomálne choroby, to znamená genómové a chromozomálne mutácie (kapitola V);
Skupina III – ochorenia s dedičnou predispozíciou, v patogenéze ktorých sa podieľajú faktory prostredia a dedičnosti, ktoré majú monogénny alebo polygénny typ dedičnosti (krátkozrakosť, morbídna obezita, žalúdočný vred).
Skupina IV – genetické ochorenia somatických buniek, často spojené s malígnymi novotvarmi (retinoblastóm, Wilmsov nádor, niektoré formy leukémie);
Skupina V – choroby genetickej inkompatibility medzi matkou a plodom, ktoré sa vyvíjajú v dôsledku imunitnej reakcie matky na antigény plodu (nekompatibilita pre Rh faktor a niektoré ďalšie erytrocytárne antigén-protilátkové systémy).
Dedičné choroby sa môžu začať prejavovať v rôznom veku. Povaha prejavu (čas objavenia sa prvých príznakov ochorenia) je špecifická pre rôzne formy dedičnej patológie. Dedičné ochorenia sú spravidla charakterizované chronickým (dlhodobým) progresívnym (so zvyšujúcou sa závažnosťou symptómov) priebehom.
9.2 Chromozomálne choroby
Do tejto skupiny patria choroby spôsobené abnormalitami v počte alebo štruktúre chromozómov. Asi 1 % novorodencov má abnormálny karyotyp a medzi mŕtvo narodenými deťmi je výskyt aberácií v počte alebo štruktúre chromozómov 20 %. Spoločné charakteristické znaky chromozomálnych ochorení sú: nízka pôrodná hmotnosť, oneskorený vývoj, nízky vzrast, mikrocefália, mikrognatia, poruchy osteogenézy, abnormálne postavenie očí. Podrobnejší popis chromozomálnych ochorení je uvedený v častiach 5.8 a 5.9.
9.3 Génové choroby
Génové ochorenia sú patologické stavy spôsobené génovými mutáciami. Najčastejšie sa tento koncept aplikuje na monogénne ochorenia.
Táto skupina sa vyznačuje heterogenitou – rovnaké ochorenia môžu byť spôsobené mutáciami v rôznych génoch. Všeobecné princípy rozvoja patológie na génovej úrovni môžu byť:
Produkcia abnormálneho proteínového produktu;
Nedostatok normálnych bielkovín;
Nedostatočné množstvo normálneho proteínu;
Prebytok normálneho proteínového produktu.
Na základe povahy porúch homeostázy (stálosti vnútorného prostredia tela) sa rozlišujú tieto skupiny génových ochorení:
1. Choroby metabolizmu aminokyselín.
Najväčšia skupina dedičných metabolických ochorení. Takmer všetky sa dedia autozomálne recesívnym spôsobom. Príčinou chorôb je nedostatok jedného alebo druhého enzýmu zodpovedného za syntézu aminokyselín.
fenylketonúria- porucha premeny fenylalanínu na tyrozín v dôsledku prudkého poklesu aktivity fenylalanínhydroxylázy - autozomálne recesívne ochorenie. Objavuje sa vo veku 2-4 mesiacov, prvými príznakmi sú letargia, kŕče, ekzémy, „myší“ pach (zápach ketónov). Postupne sa vyvíja ťažké poškodenie mozgu, čo vedie k prudkému poklesu inteligencie až idiotizmu. Ak od prvých dní života úplne vylúčite (alebo výrazne obmedzíte množstvo) fenylalanín zo stravy chorého dieťaťa pred pubertou, príznaky sa nerozvinú. Ochorenie je spôsobené mutáciami v géne PAH, ktorý kóduje fenylalanín 4-hydroxylázu. Gene PAH lokalizované na HSA12q24.1. V rôznych populáciách bolo opísaných niekoľko desiatok mutácií tohto génu. Existujú diagnostické systémy založené na PCR, ktoré dokážu odhaliť heterozygotný nosič. Nedávno boli vyvinuté nové prístupy k liečbe fenyketonúrie – substitučná liečba fenylalanín lyázou, rastlinným enzýmom, ktorý katalyzuje rozklad fenylalanínu na neškodné metabolity, a génová terapia vložením normálneho génu fenylalanín hydroxylázy do genómu.
Alkaptonúria- autozomálne recesívne ochorenie metabolizmu tyrozínu a hromadenie kyseliny homogentisovej v telesných tkanivách (kĺbová chrupavka, šľachy). Manifestácia sa vyskytuje v detstve. Prvým príznakom je stmavnutie moču. Často sa vyvíja urolitiáza a pyelonefritída. Akumulácia produktov rozkladu kyseliny homogentisovej vedie k poškodeniu kĺbov (predovšetkým kolena a bedra). Existuje tmavnutie a zvýšená krehkosť spojivového tkaniva. Charakteristické je stmavnutie skléry a uší. Mutácie v gen HGD Príčinou tohto ochorenia sú oxidázy kyseliny homogentisovej. Tento gén obsahuje 14 exónov a je lokalizovaný v HSA3q21-23. Bolo opísaných asi 100 rôznych mutácií missense, frameshift a splice site, ktoré sú spojené s touto chorobou .
Okulokutánny albinizmus 1– absencia alebo výrazný nedostatok pigmentu v koži, vlasoch, dúhovke a pigmentových membránach oka (obrázok IX, 1).
Obrázok IX, 1. Zástupcom negroidnej rasy je albín. Na základe materiálov zo stránky http://upload.wikimedia.org/wikipediacommons/99a/Albinisitic_man_portrait
Ochorenie s autozomálne recesívnym typom dedičnosti. Prejavuje sa rôznym stupňom depigmentácie kože, vlasov, dúhovky a pigmentových membrán oka, zníženou zrakovou ostrosťou, svetloplachosťou, nystagmom, častým úpalom. Rôzne missense, frameshift a nezmyselné mutácie v tyrozinázovom géne ( TYR, HSA11q24) sú zodpovedné za toto ochorenie.
2. Poruchy metabolizmu uhľohydrátov
galaktozémia- absencia alebo výrazné zníženie aktivity enzýmu galaktóza-1-fosfát-uridyltransferáza a akumulácia galaktózy a jej derivátov v krvi, ktoré majú toxický účinok na centrálny nervový systém, pečeň a očné šošovky. V prvých dňoch a týždňoch života sa pozoruje žltačka, zväčšenie pečene, nystagmus, svalová hypotónia a zvracanie. Postupom času sa vyvíja šedý zákal a retardácia fyzického a duševného vývoja. Charakterizované neznášanlivosťou mlieka.
Ochorenie má autozomálne recesívny spôsob dedičnosti. Niekoľko foriem tohto ochorenia je spôsobených rôznymi mutantnými alelami génu GALT(galaktóza-1-fosfát uridyltransferáza), lokalizovaná v oblasti HSA9p13. Missense mutácie znižujú aktivitu enzýmu v rôznej miere, čo určuje rôzne stupne závažnosti symptómov ochorenia. Napríklad Durtheho galaktozémia je takmer asymptomatická, zaznamenáva sa iba tendencia k poruchám pečene.
Gierkeho choroba (glykogenóza typu I, ochorenie ukladania glykogénu typu I)- neschopnosť premeniť glukózu-6-fosfát na glukózu, čo vedie k narušeniu syntézy a rozkladu glykogénu. Ukladanie glykogénu sa vyskytuje, ale opačný proces nie. Vyvíja sa hypoglykémia. Akumulácia nadbytočného glykogénu v pečeni a obličkách vedie k zlyhaniu pečene a obličiek. Typ dedičnosti je autozomálne recesívny. Príčinou ochorenia je mutácia v géne G6PC, ktorý kóduje enzým glukóza-6-fosfatázu. Bolo opísaných štrnásť mutantných alel tohto génu, ktoré sú spojené s Gierkeho chorobou. Existujú molekulárne genetické testy na identifikáciu heterozygotného nosičstva a prenatálna diagnostika tohto ochorenia.
3. Poruchy metabolizmu lipidov
Niemann-Pickova choroba typu A a B- znížená aktivita enzýmu lyzozomálnej kyslej sfingomyelinázy, ktorá je kódovaná génom SMPD1(HSA11p15.4-p15.1). Typ dedičnosti je autozomálne recesívny. Porušenie metabolizmu lipidov vedie k akumulácii lipidov v pečeni, pľúcach, slezine a nervových tkanivách. Charakterizované degeneráciou nervových buniek, narušením nervového systému, zvýšenou hladinou cholesterolu a lipidov v krvi. Typ A je smrteľný v ranom detstve. Pacienti typu B sú miernejší; Rôzne typy sú spôsobené rôznymi mutáciami v géne SMPD1.
Gaucherova choroba (glykozylceramidová lipidóza)- akumulácia glukocerebrozidov v bunkách nervového a retikuloendoteliálneho systému spôsobená nedostatkom enzýmu glukocerebrosidázy, ktorý je kódovaný génom GBA(HSA1q21). Patrí do skupiny lyzozomálnych akumulačných chorôb. Niektoré formy ochorenia sa prejavujú ťažkým poškodením pečene, sleziny, nervového a kostného tkaniva.
4. Dedičné choroby metabolizmu purínov a pyrimidínov
Leschov-Nychenov syndróm - recesívne ochorenie viazané na pohlavie, pri ktorom sa obsah kyseliny močovej vo všetkých telesných tekutinách prudko zvyšuje. Dôsledkom toho je oneskorenie vo vývine, stredne ťažká mentálna retardácia, ataky agresívneho správania so sebapoškodzovaním. Nedostatočná enzymatická aktivita hypoxantín-guanín fosforibozyltransferázy v dôsledku mutácií v géne HPRT1(HSAXq26-q27.2) je základom tohto ochorenia. Bolo popísaných niekoľko mutácií v tom istom géne, ktorých výsledkom je dna(zhoršený metabolizmus purínov a ukladanie zlúčenín kyseliny močovej v tkanivách).
5. Poruchy metabolizmu spojivového tkaniva
Marfanov syndróm (pavúčie prsty, arachnodaktýlia)- poškodenie spojivového tkaniva v dôsledku mutácie génu FBN1(HSA15q21.1), zodpovedný za syntézu fibrilínu. Dedí sa autozomálne dominantným spôsobom. Klinický polymorfizmus ochorenia sa vysvetľuje veľkým počtom mutantných alel, z ktorých každá sa môže prejaviť v heterozygotnom stave. Pacienti sa vyznačujú vysokým vzrastom, astenickou postavou (neúmerne dlhé končatiny), arachnodaktýliou (dlhé tenké prsty), slabosťou väzivového aparátu, odchlípením sietnice, subluxáciou šošovky, prolapsom mitrálnej chlopne (obrázok IX, 2).

Obrázok IX, 2. Marfanov syndróm. Na základe materiálov zo stránky http://www.spineinfo.ru/infosources/case/cases_14.html.
Mukopolysacharidózy- skupina ochorení spojivového tkaniva spojená s poruchou metabolizmu kyslých glykozaminoglykánov (mukopolysacharidov) spôsobených nedostatkom niektorých lyzozomálnych enzýmov. Tieto ochorenia sú klasifikované ako lyzozomálne ochorenia. Prejavujú sa rôznymi defektmi kostí a spojivového tkaniva. Mukopolysazaridóza typu I (Hurlerov syndróm) je autozomálne recesívne ochorenie vyplývajúce z deficitu enzýmu alfa-L-iduronidázy v dôsledku mutácií v géne IDUA (HSA4q16.3). To vedie k akumulácii proteínovo-sacharidových komplexov a tukov v bunkách tela. Výsledkom je, že pacienti pociťujú nízky vzrast, výraznú mentálnu retardáciu, zväčšenie pečene a sleziny, srdcové chyby, zakalené rohovky, deformácie kostí a zhrubnutie čŕt tváre (obrázok IX, 3).

Obrázok IX, 3. Hurlerov syndróm. Na základe materiálov zo stránky http://medgen.genetics.utah.edu/photographs/pages/hurler_syndrome.htm.
Mukopolysacharidóza typu II(Hunterov syndróm) je recesívne ochorenie viazané na pohlavie, ktoré je spôsobené defektom enzýmu iduronátsulfotázy v dôsledku mutácie v géne IDS (HSAXq28). Akumulačnými látkami sú dermatan a heparansulfáty. Charakterizované drsnými črtami tváre, skafocefáliou, hlučným dýchaním, nízkym hrubým hlasom, častými akútnymi respiračnými vírusovými infekciami (obrázok IX, 4 ) . Vo veku 3-4 rokov sa objavujú poruchy koordinácie pohybov - chôdza sa stáva nemotornou, deti pri chôdzi často padajú. Pacienti sa vyznačujú emočnou labilitou a agresivitou. Pozoruje sa aj progresívna strata sluchu, nodulárne kožné lézie chrbta, osteoartritída a lézie rohovky.
\ 
Obrázok IX, 4. Hunterov syndróm. Na základe materiálov zo stránky http://1nsk.ru/news/russia/23335.html.
Mukopolysacharidóza typu III (Sanfilippo syndróm, Sanfilippo choroba) - ochorenie spôsobené akumuláciou heparan sulfátu. Vyznačuje sa genetickou heterogenitou – existujú 4 typy tohto ochorenia, spôsobeného mutáciami 4 rôznych génov kódujúcich enzýmy podieľajúce sa na metabolizme nahromadených látok. Prvé príznaky ochorenia vo forme porúch spánku sa objavujú u detí starších ako 3 roky. Postupne vzniká apatia, dochádza k oneskoreniu psychomotorického vývinu, poruche reči, hrubnú črty tváre. Deti časom prestávajú spoznávať ľudí okolo seba. Pacienti sú charakterizovaní retardáciou rastu, kĺbovými kontraktúrami, hypertrichózou a stredne ťažkou hepatosplenomegáliou. Na rozdiel od Hurlerovho a Hunterovho syndrómu pri Sanfilippo chorobe prevláda mentálna retardácia a nedochádza k žiadnym léziám rohovky a kardiovaskulárneho systému.

Obrázok IX, 5. Sanfilippo syndróm. Na základe materiálov zo stránky http://runkle-science.wikispaces.com/Sanfilippo-syndrome.
Fibrodysplázia (myositis ossificans, paraosézna heterotopická osifikácia, Munheimerova choroba)- ochorenie spojivového tkaniva spojené s jeho progresívnou osifikáciou v dôsledku mutácie génu ACVR1(HSA2q23-q24), ktorý kóduje receptor aktivínu A. Typ dedičnosti je autozomálne dominantný. Ochorenie sa prejavuje vrodenými vývojovými chybami – predovšetkým zakrivenými palcami na nohách a poruchami v krčnej chrbtici na úrovni stavcov c2 – c7. Ochorenie má progresívny charakter, vedie k významným poruchám funkčného stavu pohybového aparátu, hlbokej invalidite pacientov a smrti najmä v detskom a mladom veku (obrázok IX, 6). Ochorenie sa tiež nazýva „choroba druhej kostry“, pretože tam, kde by sa v tele mali vyskytnúť normálne protizápalové procesy, začína rast kostí.

Obrázok IX, 6. Fibrodysplázia. Na základe materiálov zo stránky http://donbass.ua/news/health/2010/02/15.
6. Poruchy cirkulujúcich bielkovín
Hemoglobinopatie- dedičné poruchy syntézy hemoglobínu. Existujú dve skupiny hemoglobinopatií. Prvý je charakterizovaný zmenou primárnej štruktúry globínového proteínu, ktorá môže byť sprevádzaná poruchami jeho stability a funkcie (napr. kosáčiková anémia). Pri hemoglobinopatiách druhej skupiny zostáva štruktúra hemoglobínu normálna, znižuje sa iba rýchlosť syntézy globínových reťazcov (napr. β -talasémia).
7. Metabolické poruchy v červených krvinkách
Dedičná sférocytóza- vrodený nedostatok lipidov membrán červených krviniek. Ochorenie je charakterizované autozomálne dominantným alebo autozomálne recesívnym spôsobom dedičnosti v závislosti od génovej mutácie SPTA1(HSA1q21), ktorý kóduje erytrocytový a-1 spektrín. Anomália tohto proteínu vedie k zvýšeniu koncentrácie iónov sodíka vo vnútri erytrocytu a prenikaniu prebytočnej vody do neho v dôsledku zvýšenia osmotického tlaku. V dôsledku toho sa vytvárajú sférické červené krvinky - sférocyty, ktoré na rozdiel od bikonkávnych normálnych červených krviniek nemajú schopnosť meniť tvar v úzkych oblastiach prietoku krvi, napríklad pri prechode do slezinových dutín. To vedie k spomaleniu pohybu erytrocytov v dutinách sleziny a odlúčeniu časti erytrocytovej membrány s tvorbou mikrosférocytov. Zničené červené krvinky sú absorbované makrofágmi sleziny. Hemolýza červených krviniek vedie k hyperplázii miazgových buniek a zväčšeniu sleziny. Jedným z hlavných klinických príznakov je žltačka. Hlavnými príznakmi dedičnej sférocytózy sú zväčšená slezina (zvyčajne vyčnievajúca spod hypochondria o 2 - 3 cm) a žltačka. Niekedy sa vyskytujú známky oneskoreného vývoja, poruchy tvárového skeletu, vežovitá lebka, sedlový nos, vysoké podnebie, porušenie postavenia zubov, úzke očné jamky.
8. Dedičné choroby metabolizmu kovov
Konovalov-Wilsonova choroba (hepatocerebrálna dystrofia)– autozomálne recesívna porucha metabolizmu medi, ktorá vedie k závažnému poškodeniu centrálneho nervového systému a vnútorných orgánov. Ochorenie je spôsobené nízkou alebo abnormálnou syntézou ceruloplazmínu (proteín transportujúci meď) v dôsledku nedostatočnej enzymatickej aktivity ATPázy prenášajúcej meď. Mutácie (bolo ich opísaných asi 200) v gen ATP7B(HSA13q14-q21) vedú k zmenám v β-polypeptide tohto enzýmu, ktorý je genetickým základom tejto patológie. Hlavnú úlohu v patogenéze hrá narušenie metabolizmu medi, jej akumulácia v nervovom, obličkovom, pečeňovom tkanive a rohovke, čo vedie k toxickému poškodeniu týchto orgánov meďou. V pečeni sa tvorí veľká nodulárna alebo zmiešaná cirhóza. V obličkách sú primárne postihnuté proximálne tubuly. V mozgu sú najviac postihnuté bazálne gangliá, zubaté jadro mozočka a substantia nigra.
9. Malabsorpcia v tráviacom trakte
Cystická fibróza (cystická fibróza) - autozomálne recesívne ochorenie charakterizované poškodením exokrinných žliaz, ťažkou dysfunkciou dýchacieho systému a gastrointestinálneho traktu. Príčinou sú génové mutácie CFTR(HSA7q31.2), ktorý kóduje transmembránový regulátor cystickej fibrózy. Ochorenie je charakterizované poškodením exokrinných žliaz, ťažkou poruchou funkcie dýchacieho systému a gastrointestinálneho traktu.
Intolerancia laktózy (hypolaktázia) – autozomálne recesívny patologický stav zlej absorpcie laktózy (mliečneho cukru), ktorého genetickým základom sú mutácie v regulačných a kódujúcich oblastiach génu LCT(HSA2q21), ktorý kóduje laktázu. Tento enzým je exprimovaný prevažne v ciliovaných bunkách čreva a je zodpovedný za rozklad laktózy na galaktózu a glukózu. Hlavnými príznakmi nedostatku laktázy sú plynatosť, bolesť brucha, hnačka a vracanie. U detí sa nedostatok laktázy môže prejaviť ako chronická zápcha, nepokoj a plač po jedle. V rôznych ľudských populáciách sa frekvencia mutantných alel pohybuje od 1 do 100 %.
10. Hormonálne poruchy
Feminizácia semenníkov (Morrisov syndróm) – recesívne ochorenie viazané na pohlavie, keď mužský karyotyp (46, XY) prejavuje ženský fenotyp. Expresivita je rôzna. Pri neúplnej feminizácii sa pohlavné žľazy vyvíjajú podľa mužského typu, ale niektoré pohlavné znaky zodpovedajú ženskému pohlaviu s rôznym stupňom závažnosti - hypertrofovaný klitoris, neúplné uzavretie sutury mieška, veľké pysky ohanbia, skrátená vagína (obrázok IX, 7) . Pri úplnej feminizácii je hlavným príznakom absencia menštruácie a sexuálneho rastu vlasov s dobre vyvinutými mliečnymi žľazami a ženským fenotypom. Príčinou ochorenia sú rôzne mutácie v géne AR(HSAXq11-q12), ktorý kóduje androgénny receptor.

Obrázok IX, 7. Pohľad na vonkajšie genitálie počas neúplnej feminizácie semenníkov. Na základe materiálov zo stránky http://www.health-ua.org/img/woman/tabl/8_17.jpg.
Androgenitálny syndróm (ženský pseudohermafroditizmus) – endokrinná porucha s autozomálne recesívnym typom dedičnosti, pri ktorej má pacient mužské vonkajšie pohlavné orgány a ženskú hormonálnu štruktúru. Pacienti majú zväčšený klitoris, ktorý sa podobá mužskému penisu s jedným urogenitálnym otvorom, nemá vonkajší vchod do pošvy, chýbajú malé pysky ohanbia a veľké pysky vyzerajú ako „prerezaný“ miešok. V tomto prípade môžu mať vnútorné pohlavné orgány normálny vzhľad. Genetický základ ochorenia tvoria génové mutácie CYP21(HSA6q21.3), ktorý kóduje enzým 21-hydroxylázu skupiny cytochrómu P450, podieľajúci sa na syntéze hormónov aldosterónu a kortizolu.
9.4 Molekulárne markery pri štúdiu dedičnej patológie
Významná časť dedičných chorôb a chorôb s dedičnou predispozíciou nemá monogénny charakter. Možno ich zaradiť medzi kvantitatívne znaky, teda také, ktoré majú súvislý rozsah variability a možno ich merať – napríklad výška, hmotnosť, dĺžka končatín. Alely veľkého počtu génov prispievajú k prejavu takýchto znakov, a preto sa nazývajú polygénne. Pomocou genetických markerov je možné vysledovať ich dedičnosť a identifikovať gény, ktorých alely sa podieľajú na patologických procesoch. Identifikácia prepojenej dedičnosti (asociácia) fenotypových znakov s genetickými markermi umožňuje nájsť oblasti chromozómov, ktoré majú rozhodujúci vplyv na skúmané procesy (pozičné klonovanie) a získať spoľahlivé systémy pre molekulárnu diagnostiku (molekulárne značenie). V súčasnosti sú najbežnejšími markermi v humánnej genetike mikrosatelitné lokusy (obrázok IX, 8; časť 8.1) a mononukleotidové polymorfné miesta - SNP (obrázok IX, 9), ktorých hlavné znaky sú uvedené v tabuľke IX, 1.
Analýza génovej expresie (všetkých alebo skupiny) na biočipoch v tkanivách súvisiacich so špecifickým dedičným ochorením za normálnych a patologických podmienok často umožňuje identifikovať kandidátske gény pre skúmané ochorenie. Chromozomálna lokalizácia sekvencií DNA ovplyvňujúcich kvantitatívny znak (QTL) môže byť určená na základe spoločnej dedičnosti s niekoľkými blízko umiestnenými markermi. Ak je možné nájsť markery, ktoré obmedzujú QTL na oboch stranách, potom je možné na základe údajov o genómovej sekvencii (časť 7.7 a 8.4) zostaviť zoznam génov, ktoré sú pozičnými kandidátmi na QTL skúmanej choroby. Kombináciou analýzy expresie a štúdií asociácie chorôb s molekulárnymi markermi možno identifikovať najpravdepodobnejšie kandidátske gény – tie, ktoré sa objavia na oboch zoznamoch.
Stupeň citlivosti na určité lieky a účinnosť ich užívania sa značne líšia. Na to isté ochorenie sa liek vhodný pre konkrétneho jedinca často vyberá metódou pokus-omyl. Okrem straty času tento prístup niekedy spôsobuje nenapraviteľné poškodenie zdravia. V súčasnosti boli pre veľké množstvo liekov vyvinuté markerové systémy založené na SNP, ktoré umožňujú a priori (pred experimentom) predpovedať reakciu jednotlivého organizmu na konkrétnu chemickú látku. Asociácie jednotlivých alelických variantov DNA markerov s charakteristikami biochemických reakcií sú základom individuálnej terapie (obrázok IX, 10).

Obrázok IX, 8. V mikrosatelitných lokusoch je variačnou jednotkou skupina nukleotidov.

Obrázok IX, 9. Na mononukleotidových polymorfných miestach (SNP) je jednotkou variácie jeden nukleotid.
Tabuľka IX, 1. Porovnanie hlavných charakteristík SNP a mikrosatelitov.

Obrázok IX, 10. Princíp výberu individuálnej terapie na základe polymorfizmu mononukleotidových repetícií - SNP.
Testové otázky a úlohy pre kapitolu IX
1. Do ktorej skupiny dedičných ochorení možno zaradiť cystickú fibrózu?
2. Môže heterozygot pre génovú mutáciu SPTA1 byť dedičná sférocytóza?
3. Aké dedičné ochorenie je spôsobené hromadením heparansulfátu?
4. Prečo existujú štyri možné alely SNP?
Dodatočné čítanie ku kapitole IX
N.P. Bochkov. Klinická genetika // M.: Geotar-Med. 2002. – 457 S.