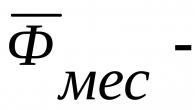7379 0
A T-sejthiányban szenvedő betegek érzékenyek a vírusos, gombás és protozoonfertőzésekre. Ezen túlmenően, mivel a T-sejtek szükségesek a T-függő antigének elleni antitestek termeléséhez, a T-sejt-hiányban szenvedő betegek az antitest-termelésben is szelektív hibákat mutatnak. Következésképpen a T-sejt-hiányban szenvedő betegeket nehéz lehet megkülönböztetni a SCID-ben szenvedő betegektől a klinikai megjelenés alapján.
Veleszületett csecsemőmirigy-aplázia (DiGeorge-szindróma)
DiGeorge szindróma egy T-sejt-hiány, amelyben a csecsemőmirigy, valamint más nem limfoid szervek rendellenesen fejlődnek. Ez a szindróma az embrionális idegi gerincsejteknek a harmadik és negyedik garattasakba történő károsodott migrációja miatt következik be. Általában ez a folyamat a terhesség 12. hetében történik. DiGeorge-szindrómában a szív, az arc, a csecsemőmirigy és a mellékpajzsmirigy rendellenességei alakulnak ki, ami thymus aplasiához és hypoparathyreosishoz vezet. A thymus aplasia következménye az érett T-sejtek hiánya és az immunhiány. A DiGeorge-szindróma modellje meztelen egerek; ezekben az állatokban nem fejlődik ki a csecsemőmirigy és a szőrtüszők.A DiGeorge-szindróma nem örökletes, hanem szórványosan fordul elő a 22q11 kromoszóma deléciója következtében. Az újszülötteknél hipokalcémiás (alacsony kalciumszint) van a mellékpajzsmirigyek hiánya és veleszületett szívhibák miatt. Az ilyen gyermekek gyakori vagy krónikus vírusos, bakteriális, gombás és protozoonfertőzésekben szenvednek. Nincsenek, vagy nagyon kevés érett T-sejtjük van a periférián (a vérben, a nyirokcsomókban vagy a lépben). Bár a B-sejtek, a plazmasejtek és a plazma Ig szintje normális lehet, sok betegnél nem tapasztalható megnövekedett antitestválasz a T-függő antigénekkel végzett immunizálás után.
Az egyéb immunglobulin izotípusokra való átálláshoz szükséges segítő T-sejtek hiánya azt eredményezi, hogy az immunizálás után hiányzik az IgG és más izotípusú antitestek. A T-független antigénekre adott IgM válasz azonban nem változik. Mivel a DiGeorge-szindrómás betegek T-sejt-hiányban szenvednek, és nem képesek normális antitestválaszt generálni, nem olthatók élő, legyengített vírusvakcinákkal!
Kezdetben a DiGeorge-szindrómás gyerekeket magzati csecsemőmirigy-transzplantációval kezelték, amelynek eredményeként egy héten belül T-sejtek jelentek meg a gazdaszervezetben. A transzplantációhoz használt magzati csecsemőmirigynek 14 hetesnél fiatalabbnak kell lennie a GVHD hiányának biztosítása érdekében, ami akkor fordulhat elő, ha érett donor csecsemőmirigy sejteket adnak egy immunhiányos recipiensnek.
A donor magzati csecsemőmirigy csecsemőmirigy-hámsejteket biztosít, amelyek lehetővé teszik, hogy a recipiens normális limfoid progenitor sejtjei T-limfocitákká fejlődjenek. Bár a keletkező T-sejtek normálisak, a sejt által közvetített immunitás és az antitest-termelésben való segítségnyújtás nem áll teljesen helyre. A recipiens T-sejtjei a transzplantált csecsemőmirigy-sejtek MHC-molekuláira „sajátjaként” emlékeznek, és néha rosszul lépnek kölcsönhatásba saját testük perifériás antigénprezentáló sejtjeivel.
Mivel ez a kezelési stratégia nem járt sikerrel, jelenleg főként tüneti terápiát alkalmaznak. Egyes betegeknél csecsemőmirigy-szövet maradványa van, amely ugyan késleltetett és csökkentett, de még mindig T-sejtek fejlődését biztosítja. A szindrómához kapcsolódó további egészségügyi probléma a veleszületett szívbetegség, amely rontja az egyébként rossz prognózist.
T-sejt-elégtelenség normál számú perifériás T-sejtekkel
Egyes betegeknél a T-sejt-hibákat inkább funkcionálisnak, mint mennyiséginek találták. Klinikailag opportunista fertőzésekkel és meglepően magas autoimmun betegségek előfordulásával járhatnak. Az ilyen családokban végzett törzskönyvek tanulmányozása autoszomális recesszív öröklődési mintát tár fel. Molekuláris vizsgálatok kimutatták a kiváltó tényezők heterogenitását, beleértve a ZAP-70, CD3ε vagy CD3ε tirozin-kináz alulexpresszióját (17.4. ábra).Rizs. 17.4. Az antigén-specifikus receptoron (T-sejt-receptor) keresztül történő jelátvitelben részt vevő molekulák hiánya
A ZAP-70 szükséges az intracelluláris jelátvitelhez, amikor a T-sejt receptorhoz kötődik. Tisztázatlan okokból a hibás ZAP-70 expresszióval rendelkező betegek SCID-szerű szindróma klinikai képét mutatják (a sejt-mediált és humorális immunitás hibáival). A T-sejt-aktivitás hiánya arra utal, hogy a ZAP-70 kritikus szerepet játszik az érett T-sejtek működésében, de a B-sejtek működésére gyakorolt hatás okai nem tisztázottak. Ezen túlmenően, bár a perifériás vérsejtszám, a nyirokcsomók és a csecsemőmirigy kezdetben normális, a hibás ZAP-70 expressziós betegekben hiányoznak a CD8+ T-sejtek.
Ez a megállapítás arra utal, hogy a ZAP-70 szükséges a CD8+ T-sejtek differenciálódásához is a csecsemőmirigyben. A CD3 láncok mutációi meglehetősen ritkák, és néhány beteget leírtak ezzel a hibával. Az állatmodellek azt sugallják, hogy CD3 peptidláncok szükségesek a normál T-sejt-receptor jelátvitelhez. Nem világos azonban, hogy az ilyen egérmodellek pontosan megfelelnek-e a korábban leírt betegeknél jelen lévő hibának.
Autoimmun limfoproliferatív szindróma
Autoimmun limfoproliferatív szindróma (ALPS)- örökletes betegség, amelyet a limfoid szövetek masszív proliferációja jellemez, a limfóma korai kialakulásával. Autoimmunnak tekinthető, mert egy ilyen genetikai hiba szisztémás autoimmun jelenséghez vezet, és csak a krónikus vírusfertőzésekkel szembeni fokozott fogékonysághoz vezet. A betegekben megnövekedett a kettős negatív (CD4 CD8) T-sejtek száma, és B-sejtes limfóma alakulhat ki. A legtöbb ALPS-ben szenvedő betegnek mutációja van a Fas fehérjét (CD95) kódoló génben.Az ezen a fehérjén keresztül továbbított jelek általában aktiválják az apoptózist vagy a programozott sejthalált. Az apoptózis aktiválása nélkül a sejtek, amelyeknek el kell pusztulniuk, élnek, és az immunválaszok, amelyeket „ki kell kapcsolni”, folytatódnak. A legtöbb autoimmun limfoproliferatív szindrómában szenvedő betegnek egy normál és egy mutáns Fas molekulája van. Ez arra utal, hogy a mutáns molekula valamilyen módon zavarja a normál Fas-molekula működését. Egyes autoimmun limfoproliferatív szindrómában szenvedő betegeknél más apoptózist kiváltó komponensek, például a Fas ligandum vagy a kaszpáz 10 hibái vannak.
Két egérfajta – az lps és a gld – fenotípusa hasonló az ALPS-ben szenvedő betegekéhez; Az lps egereknél a Fas gén, a gld egereknél pedig a Fas ligand gén mutációja van. Sok éven át tanulmányozták az lps egereket az autoimmun betegségek, különösen az SLE modelljeként, amíg fel nem fedezték a Fas génjük hibáját.
Krónikus mucocutan candidiasis
Krónikus mucocutan candidiasis a szindrómák rosszul meghatározott összessége, amelyet a bőr és a nyálkahártyák Candida nemzetségébe tartozó gombák általi fertőzése jellemez. Ezek a gombák általában jelen vannak az ilyen szövetek felületén, de nem patogének. A betegek jellemzően érintetlen sejt-közvetített immunitást mutatnak a Candidán kívül más organizmusokkal szemben, és normál B-sejt által közvetített immunitással (antitest-termelés) minden szervezettel szemben, beleértve a Candidát is. Így a T-sejtek működésében csak szelektív hiba van. Ez a betegség férfiakat és nőket egyaránt érint, gyermekkorban különösen gyakori, és bizonyos esetekben örökletes is lehet.B-sejtes vagy immunglobulinokkal összefüggő immunhiányos betegségek
A B-sejtekkel vagy immunglobulinokkal kapcsolatos immunbetegségek a hibás B-sejtfejlődéssel és az összes Ig-osztály teljes hiányával járó betegségektől az Ig egy osztályának vagy alosztályának hiányával összefüggő betegségekig terjednek. A betegek visszatérő vagy krónikus fertőzésekben szenvednek, amelyek gyermekkorban (Bruton agammaglobulinémia) vagy fiatal felnőttkorban kezdődhetnek. Ezen betegségek diagnosztizálásához szükséges a B-sejtek számának feltárása és működésük vizsgálata, az Ig osztályok és alosztályok immunelektroforetikus és kvantitatív meghatározása.X-hez kötött infantilis agammaglobulinémia
X-hez kötött infantilis agammaglobulinémia (XIA) O. Bruton írta le először 1952-ben. Ezt a betegséget Bruton agammaglobulinémiának is nevezik. Ez a rendellenesség viszonylag ritka (1 eset 100 000 emberből). Először 5-6 hónapos korban jelenik meg, amikor a baba elveszíti a placentán keresztül kapott anyai IgG-t. Ebben a korban a csecsemőknél a fő megnyilvánulások a súlyos, visszatérő bakteriális fertőzések, amelyek az összes osztályba tartozó immunglobulinok súlyos szuppressziója vagy esetleg hiánya következtében jelentkeznek.A fő hiba az, hogy a normál számban jelen lévő B-sejt-prekurzorok nem fejlődnek érett B-sejtekké. A BTK (brutoni tirozin kináz) gén, amely a CSA-ban mutációkon megy keresztül, általában egy tirozin kináz enzimet kódol, amely a citoszolban található. Úgy tűnik, hogy a fejlődő B-sejtekben elengedhetetlen résztvevője a pre-B-sejt-receptorból történő jelátviteli folyamatnak. E jel nélkül a sejtfejlődés nem folytatódik.
A mutáns gént anyától kapott egyedekben minden érett B-sejt csak egy másik aktív (nem mutáns) X kromoszóma jelenléte miatt képződik. Így a CSA egyike a számos leírt örökletes immunhiánynak, amelyeknél a citoplazmatikus tirozin kináz mutációja felelős. Korábban már leírták a JAK3 mutációit, amelyek a SCID egyik formájának kialakulását okozzák, és a ZAP-70 mutációit, amelyek felelősek a T-sejt-elégtelenség egy formájáért.
A CSA-ban szenvedő betegek vérének, csontvelőjének, lépének és nyirokcsomóinak vizsgálatai a B-sejtek és a plazmasejtek szinte teljes hiányát mutatják ki, ami megmagyarázza az alacsony Ig-szintet. Jellemző, hogy a gyermekek mandulái jelentősen fejletlenek. A korlátozott számban termelődő B-sejtek normálisan képesek plazmasejtekké válni. A CSA-ban szenvedő csecsemők visszatérő bakteriális középfülgyulladásban, hörghurutban, vérmérgezésben, tüdőgyulladásban, ízületi gyulladásban, agyhártyagyulladásban és dermatitisben szenvednek. A leggyakrabban kimutatott mikroorganizmusok a Hemophilus influenzae és a Streptococcus pneumoniae.
Gyakran a betegek felszívódási zavarban is szenvednek a Giardia lamblia dominanciája miatt a gyomor-bél traktusban. Ezenkívül az ilyen betegek érzékenyek a gyomor-bél traktuson áthatoló vírusfertőzésekre is, például az echovírusokra és a poliovírusra. A fertőzések önmagában nem reagálnak jól az antibiotikumokra, ezért a kezelés időszakos injekciókból áll intravénás immunglobulin (IVIG), amely nagy mennyiségű IgG-t tartalmaz. Bár ez a passzív immunizálás egyes betegeket 20-30 évig támogat, a prognózis óvatos, mivel a krónikus tüdőbetegségek kialakulásának valószínűsége magas a gyakori visszatérő fertőzések miatt.
Átmeneti hipogammaglobulinémia
Az 5-6 hónapos csecsemőknél a passzívan átvitt anyai IgG eltűnik, és az immunglobulinok termelése a szervezetben fokozódik. A koraszülöttek átmeneti IgG-hiányban szenvedhetnek, mivel még nem képesek immunglobulinokat szintetizálni. Alkalmanként a teljes korú csecsemőknél is csökkenhet az IgG szint, még normál IgM és IgA szint mellett is. Ez az állapot a T helper sejtek számának csökkenése és működésük megzavarása miatt következik be.Az átmeneti hypogammaglobulinémia több hónaptól 2 évig is fennállhat. Ez a betegség nem kapcsolódik a szexhez, és megkülönböztethető az X-hez kötött betegségtől a véráramban lévő normál számú B-sejt jelenléte alapján. Bár a kezelés általában nem szükséges, az ilyen csecsemőket azonosítani kell, mivel ebben az időszakban nem szabad immunizálni.
Gyakori változó immunhiány
A gyakori változó immunhiányban (CVID) szenvedő betegek szérum IgG- és IgA-szintje jelentősen csökkent, normál vagy csökkentett IgM-szinttel, valamint normális vagy csökkent B-sejtek száma a perifériás vérben. A férfiakat és nőket egyaránt érintő betegség oka nem teljesen ismert, és nem biztos, hogy minden esetben ugyanaz. A betegség bármely életkorban felléphet, de két csúcspontja van 1-5 és 15-20 éves korban. Az érintett egyéneknek visszatérő légúti és bélfertőzései vannak, amelyeket pirogén baktériumok és autoimmun betegségek, például hemolitikus anémia, thrombocytopenia és SLE okoznak, amelyek autoantitestekhez kapcsolódnak. Sokukban a sejt által közvetített immunitás zavarai is vannak. Hosszú távon az ilyen betegeknél megnövekedett a rák, különösen a limfómák és a gyomorrák előfordulása.A gyakori változó immunhiányt a B-sejtek antitest-kiválasztó sejtekké történő érésének károsodása jellemzi. Ez a hiba oka lehet a B-sejtek antigénre adott proliferációjának kudarca, vagy normál B-sejtek proliferációja IgM szekréció nélkül, vagy IgM szekréció anélkül, hogy az izotóp átváltana IgG-re vagy IgA-ra (a B- vagy T-sejtek belső károsodása miatt), vagy károsodott. nehéz IgG láncok glikozilációja. A legtöbb esetben a rendellenesség az immunglobulinok szintézisének és szekréciójának gátlása következtében nyilvánul meg. Ez a betegség családi eredetű vagy szórványosan fordul elő, és a betegséget kiváltó környezeti tényezők ismeretlenek.
A kezelés a súlyosságtól függ. A betegség súlyos formáiban, számos ismételt vagy krónikus fertőzéssel, intravénás immunglobulin kezelést írnak elő. A kezelésben részesülő betegek várható élettartama általában normális. A CVID-ben szenvedő anyák normális terhességgel rendelkeznek, de természetesen nem adják át az anyai IgG-t a magzatnak.
Szelektív immunglobulin-hiány
Számos szindrómát az immunglobulinok egy osztályának vagy alosztályának szelektív hiányosságai okoznak. Némelyikük más ellenanyag-izotípusok szintjének kompenzációs növekedésével jár együtt, például IgG- vagy IgA-hiány esetén az IgM koncentrációjának növekedése.Az IgA-hiány a leggyakoribb immunhiányos rendellenesség nyugaton, becslések szerint 800 emberből egynél fordul elő. Ennek a rendellenességnek az oka ismeretlen, de úgy tűnik, hogy összefüggésben áll az IgA B-sejtekből történő csökkent felszabadulásával. Az IgA-hiány átmenetileg is előfordulhat, a gyógyszerek mellékhatásaként. A betegek visszatérő, vírusos vagy bakteriális eredetű felső és alsó légúti fertőzésekben, cöliákiában (a bélből való felszívódás zavara) szenvedhetnek, vagy tünetmentesek.
Azon betegek kezelése, akiknél a betegség manifesztációi jelentkeznek, széles spektrumú antibiotikumok felírásából áll. A szérum immunglobulin terápiát nem alkalmazzák, mert a kereskedelmi forgalomban kapható készítmények alacsony koncentrációban tartalmaznak IgA-t, és mivel a parenterálisan beadott IgA nem éri el a szekréciós immunrendszer olyan helyeit, ahol általában védő immunglobulin. Ezenkívül a betegekben humorális válasz (általában IgG vagy IgE) alakulhat ki a transzfúziós immunszérumból származó IgA-ra, ami túlérzékenységi reakciókat okozhat. A szelektív IgA-hiány prognózisa azonban általában jó, és a legtöbb beteg jól él.
Más immunglobulin izotípusok szelektív hiányosságait is megfigyelték. Példa erre az IgM-hiány, egy ritka rendellenesség, amelyben a betegek ismétlődő és súlyos fertőzéseket tapasztalnak, amelyeket poliszacharid kapszulákat tartalmazó mikroorganizmusok, például pneumococcusok vagy Haemophilus influenzae okoznak. A szelektív IgG alosztály hiányosságait leírták, de rendkívül ritkák.
A T-B limfociták közötti interakció zavarai
Legalább két olyan betegség létezik, amelyekben a T- és B-sejtvonalak normálisan érnek, de tagjaik közötti kölcsönhatások megszakadnak. Bár mindkét állapotot a T-sejtek rendellenességei okozzák, a domináns klinikai megnyilvánulások a B-sejtekhez vagy a humorális immunválaszhoz kapcsolódnak. Ezek a betegségek a hiper-IgM szindróma és az X-hez kötött limfoproliferatív betegség.Hyper-IgM szindróma
X-hez kötött hiper-IgM szindróma (CHIM) 1-2 éves korban jelentkezik visszatérő légúti fertőzésekkel és nagyon alacsony szérum IgG-, IgA- és IgE-szinttel, normál vagy emelkedett IgM-szinttel kombinálva (lásd 17.1. ábra, 8. defektus). A B-sejtek száma az ilyen betegekben normális, in vitro teljesen működőképesek, és megfelelő stimulációval képesek az izotípusváltásra. Az X kromoszómán található CD40L4 gén mutációja a CD40 ligandum (CD154) hiányához vezet a Tn-sejteken.Ebben az esetben a CD40L a B-sejteken expresszált CD40-hez kötődik. Ez a kölcsönhatás megakadályozza a B-sejt apoptózist, és úgy tűnik, hogy fontos, ha nem elengedhetetlen, az izotípusváltáshoz. A CGIM-mel szintén nem vonzzák a B-sejteket a tüszőkhöz, ami a kialakult csíraközpontok hiányához vezet. Az ilyen rendellenességben szenvedő fiúk finom változásokat mutatnak a T-sejtek működésében és részleges blokkolást a neutrofilek differenciálódásában és a makrofágok aktiválásában. Ez magyarázhatja az opportunista fertőzésekre, különösen a Pneumocystis carinii által okozott tüdőgyulladásra való érzékenységüket, és prognózisuk rosszabb, mint a CSA-ban szenvedő betegeké.
A CGIM-hez hasonló klinikai tünetekkel rendelkező, de autoszomális recesszív típusú öröklődésű betegek második csoportjában a CD40 molekula B-sejt-hibája lehet. A harmadik csoportba tartozó betegeknél, akiknél a CD40 és az NF-κB transzkripciós faktor modulátora közötti interakció hibája, X-hez kötött öröklődési mechanizmus figyelhető meg. Amint az gyakran előfordul az intercelluláris kölcsönhatások ilyen típusú szabályozó molekuláit érintő genetikai rendellenességeknél, az érintett gyermekek olyan sejtekben is kóros elváltozásokat tapasztalnak, amelyek nem tartoznak az immunrendszerhez. Például az ilyen szabályozó molekulák sokféle sejtet használnak.
X-hez kötött limfoproliferatív rendellenesség (Duncan-szindróma)
Első X-hez kötött limfoproliferatív (XL) a betegséget hat férfi anyai rokonnál írták le a Duncan családban, így a név családi névvé vált. Ennek a ritka betegségnek a kialakulását meghatározó komplex hiba fő összetevője az, hogy a T-sejtek nem képesek szabályozni a B-sejtek növekedését. Az Epstein-Barr vírussal való érintkezés előtt az ilyen betegek klinikailag egészségesek, és normális számú T- és B-sejttel rendelkeznek. A vírussal érintkezve azonban a fertőző mononukleózis súlyos formája alakul ki, amely végzetes is lehet. Ha a beteg túléli a fertőzést, gyakran rosszindulatú limfómát vagy dysgammaglobulinémiát alakít ki.Limfóma vagy immunglobulinhiány léphet fel náluk az Epstein-Barr vírussal való előzetes érintkezés nélkül. A limfómák közül az agresszív B-sejtes limfómák túlnyomórészt a nyirokcsomókon kívül figyelhetők meg (extranodális), különösen a gyomor-bél traktusban. A leggyakoribb típus a Burkitt-limfóma. Bár a kialakuló limfómák típusa megegyezik az Epstein-Barr vírus által kiváltott, hibás T-sejt által okozott B-sejt-proliferáció szabályozásának egyéb zavaraiban szenvedő betegekével (pl. AIDS-es betegek vagy transzplantáció után immunszuppresszív kezelésben részesülő betegek), A lymphomák kialakulásának incidenciája CSL-ben szignifikánsan magasabb. A prognózis rendkívül kedvezőtlen.
A fagocita sejtek diszfunkciója
A fagocita sejtek - polimorfonukleáris leukociták és makrofágok/monociták - nagyon fontos szerepet játszanak a veleszületett és szerzett immunitásban, önmagukban vagy limfocitákkal együtt hatnak a kórokozók ellen. Az örökletes fagocita sejthiány-szindrómák segítettek azonosítani a kórokozó elpusztításához szükséges fagociták működésének minden szakaszában szükséges molekulákat.
Rizs. 17.5. (A) A károsodott sejtadhézió rontja a fehérvérsejtek azon képességét, hogy kölcsönhatásba lépjenek a vaszkuláris endotéliummal, ami zavart okoz, mivel ezek a sejtek a véráramból a fertőzés helyére vándorolnak. (B) A fagocitózishoz szükséges mechanizmusok meghibásodása a mikroorganizmusok intracelluláris pusztításának megzavarásához vezet.
Ezek a stádiumok és a hozzájuk kapcsolódó hiányosságok közé tartozik a fagocita sejtek migrációja és adhéziója (leukocita adhéziós hiány), a fagocitózis és a lizoszómákkal való fúzió (Chediak-Higashi szindróma), valamint a kórokozó elpusztítására irányuló oxidatív robbanás (krónikus granulomatózus betegség) (17.5. ábra). A fagocita diszfunkció másodlagos is lehet, amelyet külső tényezők, például gyógyszerek és szisztémás betegségek (például cukorbetegség) vagy az immunrendszer más részeinek hibái okozhatnak.
Leukocita adhéziós hiány
Ahhoz, hogy a fehérvérsejtek megérkezzenek a szövetfertőzés helyére, a sejteknek először el kell hagyniuk a véráramot. Ez a folyamat több szakaszból áll. Az első szakaszban a leukociták lassan gördülnek az endotéliumon az endothelsejteken lévő szelektinek és a leukocitákon lévő szelektin ligandumok kölcsönhatása miatt. Ekkor a kemoattraktánsok hatására a sejt mozgása (gördülése) leáll. Az ilyen sejt szorosabban kötődik, majd elkezd vándorolni az endotéliumon keresztül. Az utolsó szakasz a leukocitákon lévő integrinek és az endotélsejteken lévő ligandumaik kölcsönhatását igényli.Leukocita adhéziós hiány (LAD)- ez egy olyan rendellenességcsoport, amelyben a leukociták kölcsönhatása a vaszkuláris endotéliummal megszakad (17.5. ábra, A). Az NLA I egy autoszomális recesszív betegség, amelynek génje a 21. kromoszómához van térképezve. A betegeknél az integrin molekulák β-alegységének hibáját diagnosztizálják, ami megakadályozza azok expresszióját. Ezenkívül a β-alegység három, granulocitákon, monocitákon és limfocitákon expresszálódó integrinben közös: LFA-1 (CD1 la/CD18), Mac-1 (CD1 lb/CD18) és p150.95 (CD1 lc/CD18). . Ennek eredményeként a leukociták összes típusának adhéziója és migrációja megszakad.
Az NPA I-ben szenvedő betegek visszatérő bakteriális lágyszöveti fertőzésekben szenvednek, amelyekben megnő a fehérvérsejtszámuk, de nem termelnek gennyet, vagy nem gyógyulnak hatékonyan. A limfociták funkciói szintén károsodnak az LFA-1 expressziójának hiánya miatt. Az NLA I-ben szenvedő újszülöttek megkülönböztető jellemzője a köldökzsinór-maradvány késői elvesztése.
Az NLA II-ben szenvedő betegek szelektin ligandumainak hibája van, így sejtjeik nem tudnak végiggurulni az endothel felületén (a migráció első lépése) (lásd 17.5. ábra, A). Az NLA II kialakulását meghatározó hiba a fukóz metabolizmusával függ össze, ami a fukozilált ligandumok hiányához vezet, amelyekhez a szelektinek kötődhetnének. Az immunhiány tünetei ebben a rendellenességben kevésbé kifejezettek. Rajtuk kívül a fukóz anyagcsere hibája más fejlődési hibákhoz vezet. Az NPA I-hez hasonlóan jellemző a genny hiánya vagy jelentéktelen képződése; Ezek a gyermekek súlyos fertőzések esetén nem mutatják a gyulladás klasszikus klinikai tüneteit.
Chediak-Higashi szindróma
A Chediak-Higashi-szindróma egy autoszomális recesszív rendellenesség, amelyet a sejtekben található abnormális óriási szemcsék és organellumok jellemeznek (17.5. ábra, B). Főleg a lizoszómák és a melanoszómák érintettek, ami pigmentációs hibákhoz és a neutrofilek, NK-sejtek és vérlemezkék működési zavarához, valamint neurológiai rendellenességekhez vezet. A neutrofilek csökkent képességgel rendelkeznek az organizmusok intracelluláris elpusztítására, ami mind a hibás degranulációnak, mind a lizoszómák és a fagoszómák károsodott fúziójának az eredménye. Idővel a betegekben limfociták és makrofágok tömeges beszűrődése alakul ki a májban, a lépben és a nyirokcsomókban. A piogén mikroorganizmusok, például a streptococcusok és a staphylococcusok ismétlődő, néha végzetes fertőzéseket okoznak. A prognózis kedvezőtlen.Krónikus granulomatosus betegség
Nál nél krónikus granulomatózisos betegség (CGD) az abszorbeált organizmusok pusztításának utolsó szakasza megszakad (lásd 17.5. ábra, B). Ebben az esetben az organizmusok intracelluláris perzisztenciája granulomák kialakulásához vezet. Normális egyénekben az aktivált neutrofilek és mononukleáris fagociták elpusztítják a mikroorganizmusokat egy oxidatív kitöréssel, amely oxigént fogyaszt, és hidrogén-peroxidot és szabad szuperoxid gyököket szabadít fel. A kitörést katalizáló enzim, a NADPH-oxidáz bármelyik alegységében bekövetkező mutációk CGD-hez vezethetnek.A CGD leggyakoribb formája a membránhoz kapcsolódó alegységek, a gp91phox mutációjának köszönhető, amelyet az X kromoszómán található SUVV gén kódol. Így a legtöbb beteg X-hez kötött recesszív öröklődési mintázattal rendelkezik. A NADP oxidáz más alegységeit autoszomális gének kódolják. Az ilyen autoszomális alegységek mutációi által okozott CGD-ben szenvedő betegek autoszomális recesszív öröklődési mintázatot mutatnak. Főleg az enzim két citoszol alegységének egyikében, a p47phoxban vagy a p67phoxban vannak mutációk.
A betegség tünetei az élet első 2 évében jelentkeznek. A betegek fokozott érzékenységet mutatnak az általában alacsony virulenciájú mikroorganizmusok, például a Staphylococcus aureus, a Serratia marcescens és az Aspergillus által okozott fertőzésekre.
A kapcsolódó rendellenességek közé tartozik a lymphadenopathia (megnagyobbodott nyirokcsomók) és a hepatosplenomegalia (megnagyobbodott máj és lép), amelyek krónikus és akut fertőzésekhez kapcsolódnak. A kezelés agresszív immunizálásból és széles spektrumú antibiotikumokkal, gombaellenes szerekkel és interferon-γ-val végzett terápiából áll.
A CGD mellett azok a rendellenességek, amelyekben a fagocitózis folyamatában részt vevő enzimek aktivitása csökken vagy hiányzik, az intracelluláris organizmusok elpusztításának képességének csökkenéséhez vezetnek. Ezek közé tartozik a glükóz-6-foszfát-dehidrogenáz, a mieloperoxidáz és az alkalikus foszfatáz.
Interferon-y receptor hiány
Az IFNγR1 gén mutációja azt eredményezi, hogy a monociták nem tudnak reagálni az IFNγ-ra a tumornekrózis faktor a (TNFα) kiválasztásával. Az ezzel a mutációval rendelkező betegek szelektíven érzékenyek az enyhén patogén mikobaktériumokra, ami megerősíti az IFNy fontosságát a mikobakteriális fertőzések leküzdésében. Ez a szelektivitás arra is utal, hogy más IFNy-aktivitások kompenzálódnak az ilyen rendellenességben szenvedő egyénekben. A világ egyes részein elterjedt BCG immunizálás veszélyes lehet az ilyen hibában szenvedő betegek számára.NK sejt hiány
Nagyon keveset tudunk az emberek természetes ölősejt-hiányáról; Ennek a betegségnek csak néhány esetét írták le. Állatkísérletek azt sugallják, hogy az NK-sejt-hiány rontja az allograft kilökődést, és a vírusos betegségekre való fokozott fogékonysággal és a tumormetasztázisok megnövekedett előfordulásával jár. Az NK-sejtek defektusai súlyos kombinált immunhiányos rendellenességekben, a T-sejtek és a fagocitasejtek működésének egyes rendellenességeiben, valamint az X-hez kötött limfoproliferatív szindrómában fordulnak elő.A komplementrendszer rendellenességei által okozott betegségek
A komplement szükséges a baktériumok és a sérült sejtek opszonizálásához és elpusztításához, a kemotaxishoz és a B-sejtek aktiválásához. A komplement komponensek részt vesznek az antigén-antitest komplexek elpusztításában is, megakadályozva az immunkomplexek lerakódását és az azt követő betegségeket. A komplementhiány autoszomális tulajdonságként örökölhető, a heterozigóta egyedekben egy adott komplement komponens normál koncentrációja a fele. A legtöbb összetevő esetében ez elegendő a betegség klinikai megnyilvánulásainak megelőzésére. Az aktivált komplement komponensek felezési idejét általában szigorúan szabályozzák olyan inhibitorok, amelyek elpusztítják az aktivációs termékeket vagy disszociálják a komplexeket.A korai aktivációs fázis komplement hiánya
A korai aktivációs fázisú komplement komponensek különösen fontosak az opsonin C3b képződésében (17.6. ábra). Azoknál a betegeknél, akiknél a C1, 4 vagy 2 hiányosság a klasszikus aktivációs útvonal mentén, vagy maga a C3 hiány, gyakrabban fordul elő kapszulázott mikroorganizmusokkal (Streptococcus pneumoniae, Streptococcus pyogenes, Haemophilus influenzae) való fertőzés, valamint csökkent immunkomplexitás miatti reumás betegségek. a C3b elégtelen képződése miatt. Ennek legszembetűnőbb következménye az autoimmun betegségek gyakori előfordulása. Valójában az SLE bizonyos komplement komponensek hiányának egyik leggyakoribb megnyilvánulása.Ezeknél a betegeknél az SLE nagyon korán kezdődik, és súlyosabb, mint az SLE, amely nem jár komplementhiánnyal. Ezenkívül az ilyen SLE kialakulhat más esetekben gyakran előforduló antitestek hiányában is. A mannózkötő lektin hiánya, amely antitestek részvétele nélkül kötődik a mikrobák felületéhez, és a komplement aktiváció klasszikus útját váltja ki, szintén növeli a bakteriális fertőzések és a lupuszszerű tünetek kockázatát. Mivel a komplement aktiváció minden útja - klasszikus, lektin és alternatív - megköveteli a C3 aktiválását, maga a C3 hiánya a legsúlyosabb tünetekkel, különösen a fertőző betegségek bonyolult lefolyásával jár.
Késői aktiválási fázis komplement hiány
Ha a késői aktivációs fázis komplement komponensei (C5-C9) hiányoznak, a membrán támadó komplex (MAC) képződése megszakad. Ez a komplex a Gram-negatív baktériumok, különösen a Neisseria meningitidis közvetlen pusztítója, és az ellenük való védekezés első vonala (lásd 17.6. ábra).A komplement komponensek szabályozásának zavara
Örökletes angioödéma. Az örökletes angioödémában szenvedő betegeknél nincs hatékony C1-észteráz inhibitor. Ha ez az inhibitor hiányzik, a C1 hatása a C4-re, C2-re és a kallikrein rendszerre nem szabályozott, ami nagy mennyiségű vazoaktív peptid képződését eredményezi.
Rizs. 17.6. A komplement aktivációs kaszkád azt mutatja, hogy ha a korai aktiválási fázis komplement komponensei hiányosak, a betegek hajlamosak a kapszulázott mikroorganizmusok és a rheumatoid szindrómák által okozott fertőzésekre. A késői aktivációs fázis komplement hiánya a Neisseria fertőzésekkel jár
Ezek a peptidek növelik a vérerek permeabilitását. A betegek helyi duzzanatot tapasztalnak, amely életveszélyes lehet, ha a gégeben alakul ki, és elzárja a légutakat. A kezelés a kiváltó okok megszüntetéséből áll, amelyek általában sérüléseket is tartalmaznak, és egy C1-észteráz inhibitor infúzióját adják.
A glikozilált foszfatidil-inozithoz kapcsolódó fehérjék hiánya. A GPI-vel lehorgonyzott fehérjecsalád a vörösvérsejtek, limfociták, granulociták, endotélsejtek (az ereket bélelő sejtek) és hámsejtek membránján expresszálódik. Ezek a fehérjék, amelyek magukban foglalják a disszociációt gyorsító faktort (CD55) és a CD59-et, megvédik a sejteket a komplement általi spontán lízistől. Ha a sejtfelszínen hiányoznak az inhibitorok, a granulociták, a vérlemezkék és különösen az eritrociták érzékenyek a komplement általi spontán lízisre. Csak néhány családban van öröklött mutáció a FUD-ban, a CD59-ben vagy az összes GPI-fehérjében. Ezek a betegek súlyos vérszegénység, trombózis és krónikus fertőzések tüneteit mutatják.
Ennek a betegségnek a szerzett formája, az ún paroxizmális éjszakai hemoglobinuria (PNH), sokkal gyakrabban fordul elő. PNH-ban a betegek nem rendelkeznek az összes GPI-vel rögzített fehérje előállításához szükséges enzimmel. Egy korai mieloid progenitor sejtben szerzett szomatikus mutáció miatt fordul elő. Három nem limfoid vonal érintett: granulociták, vérlemezkék és eritrociták.
A legtöbb betegben az ezzel a mutációval rendelkező őssejtek további mutációkat szereznek, majd elkezdik uralni a normál sejteket a csontvelőben, és leállnak az érésben, ami akut mieloid leukémiához vezet. A PNH krónikus lefolyásában intravaszkuláris hemolízis alakul ki, különösen éjszaka a vesékben, ahol a savas környezet az alternatív útvonal mentén komplement aktivációt vált ki. Ez a klinikai megnyilvánulás tükröződik a betegség nevében.
Másodlagos immunhiányos betegségek
A másodlagos immunhiányos betegségek más betegségek következményei. A rossz táplálkozás (vagy annak hiánya) továbbra is az immunhiányos rendellenességek leggyakoribb oka világszerte. A fejlett országokban gyakoribb az iatrogén immunhiány, i.e. gyógyszeres terápia okozta, különösen kemoterápiás szerek rák kezelésében történő alkalmazása vagy szervátültetés vagy autoimmun betegségek esetén kontrollált immunszuppresszió következtében.Másodlagos immunhiányok is kialakulnak autoimmun betegségek esetén (nem a kezelés következményeként), vagy súlyos bakteriális fertőzésekből való felépülés után. Az immunrendszer rosszindulatú daganatai is gyakran elnyomják a normál (nem rosszindulatú) komponenseket, ami az ilyen betegek fokozott fogékonyságát eredményezi a fertőző betegségekre.
R. Koiko, D. Sunshine, E. Benjamini
A csecsemőmirigy, az immunrendszer fő szervének szerepét csak 1961-ben határozták meg R. A. Good és J. F. Miller adatainak köszönhetően. Annak ellenére, hogy számos adat áll rendelkezésre a csecsemőmirigynek a limfoid rendszer érésében betöltött szerepéről, a probléma számos aspektusa, különösen az éretlen, csecsemőmirigy előtti limfoid sejtek átalakulása jelentős funkcionális heterogenitású immunkompetens T-sejtekké, még nem teljesen tisztázott. .
A csecsemőmirigyen átjutott T-sejtek képesek reagálni az antigénekre és mitogénekre (PHA és ConA), megtelepednek a nyirokszervek csecsemőmirigy-függő zónáiban (a nyirokcsomók paracorticalis zónáiban, a lép periartériás tüszőiben) és a perifériás vérben keringő medence nagy részét alkotják hosszú élettartamú ciklussal. Ezeket a sejteket csecsemőmirigy-függőnek nevezik.
T-sejtek fejlődése
Az intratímiás fejlődést különböző tényezők biztosítják: a csecsemőmirigy stromája, valamint hormonjai, hám- és mesenchymalis sejtjei által meghatározott sajátos „mikrokörnyezet”. A csecsemőmirigyből számos hatóanyagot izolálnak, amelyek aktivitásukban, molekulatömegükben és hőmérsékleti hatásokkal szembeni ellenállásban különböznek egymástól. Közülük a timozint, a timopoietint és a thymus humorális faktort írják le. Ezek az anyagok, különösen a timozin, elősegítik a T-sejtek érettebb formákká történő átalakulását.
A timozin és más hatóanyagok T-sejtekre gyakorolt hatásmechanizmusa nem teljesen világos, úgy vélik, hogy alkalmazásuk a sejtmembrán, nevezetesen az adenilát-cikláz rendszer. Valószínűleg a timozin aktiválja, és növeli az intracelluláris cAMP és cGMP koncentrációját. Ezek a nukleotidok a sejtfejlődés folyamataihoz kapcsolódnak. Ez utóbbiak nagyon összetettek, sokrétűek és további tanulmányozást igényelnek.
A T-sejtek típusai és funkciói
A T-sejtek között több alpopulációt különböztetnek meg, amelyek különböznek a kortizol érzékenységben, valamint a felszínükön lévő antigének és receptorok különbségeiben.
Céljuk szerint a T-sejtek elsősorban a sejtes immunitásért felelősek, funkciójukban azonban heterogének. Több alosztályból állnak: antigént felismerő sejtek; segítő sejtek; gyilkos sejtek; szupresszor sejtek; memóriasejtek stb. Az a kérdés, hogy a T-sejteket mikor osztják fel különböző funkcionális célú alpopulációkra, nagyon összetett és ellentmondásos. Nem világos, hogy mi határozza meg a fejlődés funkcionális irányát, ezek az alpopulációk egyetlen prekurzorból származnak, vagy a különbség genetikailag az őssejt szintjén meghatározott.
Az összes T-sejt-alcsoport az összes limfocita körülbelül 60-70%-át teszi ki; körülbelül 25%-a B-sejt vagy bursa-dependens sejtek, amelyek az immunitás egy másik központi szervében – a Fabricius bursában (madarakban) vagy analógjaiban – „kiképzésen” estek át, ahol a T-sejtekre jellemző tulajdonságokat sajátítanak el. Meg kell jegyezni, hogy ennek az analógnak a hosszú keresése nem vezetett sikerhez. Ezt a szerepet különféle limfoid képződmények - a függelék, a mandulák, a belek csoportos nyiroktüszői, a tüdőben és más szervekben található limfoid felhalmozódások - jelölik ki. Az érést követően a B-limfociták főként a lép tüszőibe és nyirokcsomóiba költöznek.
A legtöbb antigén csecsemőmirigy-függő, mivel a rájuk adott immunválasz megköveteli a T-sejtek részvételét; ezek az antigének összetettek, több, eltérő specificitású (szöveti, mikrobiális, vírusos) antigéndeterminánssal rendelkeznek.
A T-sejtek antigénfelismerésének mechanizmusa nagyon összetett, és nem vizsgálták teljesen. A T-sejtek és receptoraik fő feladata az „én” és a „nem-én” felismerése. antigének. Az antigéneket felismerő T-sejt-receptorok etiológiáját nem határozták meg véglegesen. A sejtreceptorok és az antigén kölcsönhatása az immunválasz és a sejtdifferenciálódás megindításának jele. Ha az immunválasz az antitestképződés típusát követi, akkor a B-sejtek részvételéhez és plazmasejtekké való átalakulásához a helper T-sejtek (Th) részvétele szükséges. A T-sejtek által végrehajtott sejtes reakciók kialakulása asszisztensek - T-erősítők (Ta) részvételével is megtörténik.
A T-limfociták segítő hatásának mechanizmusa (közvetlen érintkezés, vagy a sejtek által kiválasztott hatóanyagok) nagyon összetett és nem teljesen ismert. Az aktivált Th specifikus faktorokat termel, és makrofágokon keresztül aktiválja a B-sejteket. E tényezők lényegét még nem határozták meg teljesen. Figyelembe véve, hogy a B-sejtek különböző osztályú immunglobulinokat termelnek, lehetséges, hogy különböző Th-szubpopulációk léteznek az antitestképződés szabályozására (IgG, IgE).
Gershon 1969-ben dolgozta ki a T-sejtek (T-szupresszorok) szuppresszív szerepének koncepcióját, majd kiderült, hogy a T-k képesek az immunválasz különböző fázisait szabályozni. A szuppresszorok szerepe az, hogy szabályozzák a specifikus immunválasz szintjét és erősségét, biztosítva az immunológiai toleranciát egyes csecsemőmirigy-függő antigénekkel és szöveteik antigénjeivel szemben. A T-k IgG, IgM receptorokat hordoznak, és oldható mediátorokon keresztül hatnak. A Ts által kiválasztott faktorok természetét aktívan tanulmányozzák. Nincs konszenzus a Ts indukció mechanizmusairól (a sejtek közvetlen érintkezése antigénnel vagy visszacsatolási jelekkel).
A Ts értéke nagyon magas az immunválasz korlátozása nem kevésbé fontos a szervezet számára, mint annak stimulálása. Ez különösen vonatkozik azokra az immunreakciókra, amelyek patológiához vezethetnek (autoimmun betegségek, betegségek)
A gyilkos T-sejtek celluláris immunreakciókat hajtanak végre: citopatogén hatások a transzplantációs immunitásban, daganatellenes immunitásban és bizonyos típusú fertőzésekben. A gyilkos sejtek egy szubpopulációja a késleltetett típusú túlérzékenység (DTH) effektora is.
A T-sejt populáció heterogenitása funkcionális tulajdonságaik tekintetében megmagyarázza a sejtek rendkívül összetett kölcsönhatását a szervezet antigénre adott reakcióiban.
Ma már nyilvánvaló, hogy nemcsak a T-sejtek, hanem a B-sejtek sem alkotnak homogén sejttömeget. A B-sejteket immunglobulinok jelenléte jellemzi, amelyek biztosítják az antigének felismerését, amelyek hatására a B-limfociták plazmasejtekké alakulnak, amelyek antitesteket termelnek -. A legtöbb antigén termelődése nem mehet végbe T-sejtekkel való kölcsönhatás nélkül. A sejtek együttműködése elengedhetetlen feltétele az immunválasz kialakulásának.
Az aktivált T-sejtek a Ts és Th által szekretált faktorokon kívül számos mediátort - limfokineket - képeznek, amelyek eltérő fizikai-kémiai tulajdonságokkal rendelkeznek. Szerepük különböző sejtek – neutrofil granulociták, makrofágok, eozinofil granulociták – részvétele az immunválaszban. Ezeknek a T-sejteknek a részvétele az immunválaszban nagyon fontos. Különösen fontos a makrofágok szerepe, amelyek az antigént elpusztítják és immunogén formává alakítják. A makrofágok jelenleg fagocitózisra képes, üveghez tapadó sejtek csoportját foglalják magukban – adhereneket, fagocita mononukleáris sejteket, szöveti makrofágokat és monocitákat.
A cikket készítette és szerkesztette: sebészFelnőttkori T-sejtes leukémia
A felnőttkori T-sejtes leukémia vírus endémiás, vagyis csak a világ bizonyos régióiban terjed az emberek között. A legtöbb Japánban és a karibi szigeteken található. Magas szintű fertőzést találtak Pápua Új-Guinea, Ausztrália és a Salamon-szigetek őslakosai körében. A vírus másik góca a Kaszpi-tenger környékén alakult ki. Oroszországban a vírus meglepő módon csak a Szahalin-sziget központi részén található Nogliki faluban található nivkheknél. A vírus emberről emberre terjed a szoptatás, a nemi érintkezés és a szennyezett vér átömlesztése során (vagy ha a kábítószer-függők közös fecskendőt használnak).
Az endémiás területeken sok ember megfertőződik a vírussal, de általában a fertőzött egyének életük végéig tünetmentes vírushordozókká válnak. Csak a hordozók 2-3%-ánál szegi meg a vírus egy hosszú, évtizedes látens időszak után a „hallgatási fogadalmat”. Ebben az esetben rosszindulatú betegség alakul ki, amelyben az éretlen limfociták száma meredeken megnő, a máj és a lép megnagyobbodik, a csontszövet elpusztul, és gyakran bőrkiütés alakul ki.
A T-sejtes leukémia vírus célpontja a T-limfociták. A fertőzés után a vírus beépíti genetikai anyagát a gazda kromoszómájába. Bár a vírusnak nincs saját onkogénje, a vírusfehérjék nagyszámú sejtgént aktiválnak, beleértve a sejtes onkogéneket is. Így a fertőzött sejtek, amelyekben a vírus hirtelen aktiválódik, rosszindulatúvá válnak, és ellenőrizhetetlenül szaporodni kezdenek. Ezenkívül fokozzák az interleukinok szintézisét irányító gének munkáját - olyan kis fehérjéket, amelyek segítségével az immunrendszer sejtjei kommunikálnak egymással. A T-sejtes leukémia vírus által kiváltott interleukinek számának meredek növekedése információs zajt kelt, amely szétzilálja az immunrendszer működését. Különösen a gyilkos T-sejtek száma csökken. Az immunrendszert megfosztják a daganatsejtek legyőzésének legfontosabb eszközeitől, és már nem tud megbirkózni azok terjeszkedésével. Ezért ennek a betegségnek a prognózisa rossz: a várható élettartam általában nem haladja meg a diagnózist követő hat hónapot.
A felnőttkori T-sejtes leukémia vírus az ember egyik legősibb kísérője. Úgy gondolják, hogy körülbelül 20 ezer évvel ezelőtt keletkezett. A dél-amerikai indiánok és az afrikai pigmeusok, vagyis a külvilágtól hosszú ideig elszigetelt törzsek képviselői között találták meg. A vírus genetikai sokféleségének tanulmányozása lehetővé teszi, hogy nyomon kövessük az ősi emberek vándorlási útvonalait. Különösen a T-sejtes leukémia vírus ázsiai és amerikai izolátumainak összehasonlítása szolgáltatott további bizonyítékot arra a hipotézisre, hogy az amerikai indiánok ősei ázsiai eredetű mongoloidok voltak. Valószínűleg 10-40 ezer évvel ezelőtt az Ázsiát és Északot összekötő földszoros mentén hatoltak be Amerikába.
Amerika a jelenlegi Bering-szoros helyén, és az egész amerikai kontinensen letelepedett.
A szerző Great Soviet Encyclopedia (CL) című könyvéből TSB A szerző Great Soviet Encyclopedia (LE) című könyvéből TSB A szerző Great Soviet Encyclopedia (SHK) című könyvéből TSB Emily Post Encyclopedia of Etiquette című könyvéből. A jó modor és a kifinomult modor szabályai minden alkalomra. [Etikett] írta Peggy's Post A Biológia című könyvből [Teljes kézikönyv az egységes államvizsgára való felkészüléshez] szerző Lerner György IsaakovichFELNŐTT SZÜLETÉSNAPOK A születésnapos fiú házastársai, gyermekei és olykor közeli barátai ünnepélyesen meg akarják ünnepelni valamelyik „kerek” dátumát, például a 30., 40. vagy 50. évfordulót. Ebben a kérdésben nincs különösebb szabály vagy korlátozás. Egy ilyen esemény megünneplése
A Fonott bútorok című könyvből írta: Antonov E1.3. Az élőtermészet fő szerveződési szintjei: sejtes, szervezeti, populációs-faji, biogeocenotikus A vizsgadolgozatokban tesztelt alapfogalmak: életszínvonal, ezen a szinten vizsgált biológiai rendszerek,
A Complete Medical Diagnostics Guide című könyvből szerző Vyatkina P. Az Atlasz: emberi anatómia és élettan című könyvből. Teljes gyakorlati útmutató szerző Zigalova Elena JurjevnaLeukémia Akut és krónikus leukémiában szenvedő betegeknél gyengeség, letargia és rossz közérzet figyelhető meg. Az akut leukémiák közé tartoznak a vérrendszer daganatos megbetegedései, amelyek fő szubsztrátja a blastsejtek: myeoloblastok, limfoblasztok, monoblasztok, eritroblasztok,
Egy fiatal háziasszony teljes enciklopédiája című könyvből szerző Polivalina Ljubov AlekszandrovnaLeukémia Felnőtteknél akut myeloblastos, limfoblasztos és minden egyéb akut leukémia esetén a VAMP-kúra hatékony (8 napos kúra: metotrexát - 20 mg/m2 intravénásan az 1. és 4. napon, vinkrisztin - 2 mg/m2 naponta a 2. napon Az intravénás kezelés 1. napja, 6-merkaptopurin -
Az Encyclopedia of Early Development Methods című könyvből szerző Rapoport Anna A Fotelek, székek, asztalok, polcok és egyéb fonott bútorok című könyvből szerző Podolsky Jurij FedorovicsJátékok felnőtteknek Egy nyaralás nem ünnep, ha triviális italozássá válik. És hogy ez ne forduljon elő, azt tanácsoljuk, háziasszonyok, hogy ne feledkezzenek meg a közvetlenül az asztalnál játszható játékokról és szórakoztatásról „Húzza ki a választ” Ez nagyon egyszerű és egyben
A tolószekrények, folyosók, csúszdák, falak, polcok, komódok és egyéb előregyártott bútorok könyvből szerző Podolsky Jurij Fedorovics Dr. Myasnikov Encyclopedia című könyvéből a legfontosabb dolgokról szerző Myasnikov Alekszandr Leonidovics A legteljesebb baromfitenyésztői útmutató című könyvből szerző Slutsky Igor A szerző könyvéből5.4. Felnőttek védőoltása Minden felnőttnek, különösen a fogamzóképes korú nőknek meg kell győződnie arról, hogy megkapta a mumpsz, rubeola és kanyaró elleni védőoltást. Ha a szovjet időkben minimálisra csökkentették a kanyarót, most learatjuk a következményeit
A T-sejtes limfóma a nem-Hodgkin-rákos patológiák csoportjába tartozik, amelyek a nyirokrendszert érintik. Ez a hematológiai betegség elsősorban az idősebb férfiakat érinti, de előfordulhat nőknél is. A betegség epidernotróp eredetű, és agresszív fejlődés jellemzi.
A non-Hodgkin-betegségek osztályozása
Az emberi nyirokrendszer a fertőző betegségek elleni védekezés fő eszköze. A vírusok elleni küzdelem fő asszisztensei a limfociták.
Ezek a vérelemek három típusra oszthatók:
A non-Hodgkin-betegségek ezen sejtek módosulása miatt alakulnak ki, amelyek képesek gyorsan mutálni és szaporodni. A betegség kialakulásában szerepet játszó leukociták nevétől függően a limfómákat NK-, B- és T-sejtes daganatokra osztják.
A B-sejtes non-Hodgkin neoplazmák közül a leggyakoribb patológia:
- köpenysejtes limfóma;
- follikuláris daganat;
- plazmacitóma;
- neoplazma a marginális zónában;
- kissejtes limfóma.
Az NK sejt tumorok atipikus NK leukocitákból képződnek. A T-sejt-mutációk által okozott neoplazmák a következők:
Különböző szerkezetű és szerkezetű betegségekről van szó, amelyekre a limforetikuláris szövetekben a rosszindulatú sejtek szaporodása jellemző. A kóros folyamat kiterjed a csontvelőre, a nyirokcsomókra, az emésztőrendszerre, a májra és a lépre. A non-Hodgkin-daganatok sokkal gyakoribbak, mint a Hodgkin-kór.
A T-sejtes limfómák jellemzői
A betegség kialakulásától függően indolens és agresszív non-Hodgkin limfómákat különböztetnek meg. Az álszöveti betegség szintén rendkívül ritka. Ez a betegség hasonlít a rákos daganatokhoz, de hematológiai természetű neoplazma.
Az indolens patológiák több altípusra oszthatók:

Az indolens daganatok lassú fejlődésű passzív neoplazmák. Az agresszív limfómák növekedésük intenzitásában különböznek. Ez a betegség magában foglalja:
- Sezary szindróma;
- agresszív és primer perifériás bőr T-sejtes limfóma;
- a limfóma előzetes;
- extranodális daganat;
- felnőttkori leukémia.
Néha a passzív daganatok limfómákká alakulhatnak a betegség agresszív lefolyásával. Más esetekben a T-sejtes patológia átlagos sebességgel fejlődik.
A patológia okai
A nyirokrendszerben bekövetkezett ilyen változások okait nem sikerült teljesen azonosítani. A tudósok úgy vélik, hogy a rosszindulatú T-leukocita daganatok kialakulásának fő tényezője az 1-es típusú leukémiás humán T-sejt vírus. Alapvetően több ok is befolyásolja a T-limfómák kialakulását:
- örökletes genetikai hajlam;
- a vegyszerek, az ultraibolya és a sugárzás hosszú távú hatása a testre;
- gyulladásos folyamatok a szervezetben;
- öröklött immunhiány.
Az idősek veszélyben vannak. Mindezen tényezők és az állandó stressz, a test túlterheltsége és a rossz táplálkozás kombinációja a szöveti sejtekben változásokhoz vezethet. Az eredmény a bőr T-sejtes limfómái vagy sejtes perifériás neoplazmák kialakulása.
A betegség tünetei és diagnózisa
A fejlődés jelei szerint a sejtes T-limfóma 4 stádiumban van:
- A patológia a nyirokcsomóknak csak egy területét érinti.
- A patológia csak a rekeszizom egyik oldalán fordul elő a szellemi csomópontokban.
- A rekeszizom kétoldali károsodása.
- A módosított sejtek növekednek és elterjednek a nyirokrendszerben, és hatással vannak a létfontosságú emberi szervekre.
A negyedik stádiumú betegség áttéteket képezhet a májban, a gyomorban, a vesében és a csontvelőben. A betegség gyakran súlyosbodott, előrehaladott patológia eredményeként jelenik meg.
A T-sejt módosulás tünetei különbözőek lehetnek:
- magas fokú izzadás;
- hirtelen és hosszan tartó fogyás;
- emésztési problémák;
- általános gyengeség a testben, ingerlékenység és álmosság;
- a testhőmérséklet változásai a normától való eltérésekkel egyik vagy másik irányban.
Bőr limfómák esetén a T-sejtekben különböző formájú csomók, foltok, kiütések jelennek meg. 
Ha a leukociták változásának jeleit észleli, azonnal forduljon orvoshoz. Az onkológiai központban egy onkológus kezdeti vizsgálatot végeznek.
A betegség diagnosztizálásának következő szakasza a betegség teljes morfológiája. Szükséges vizeletvizsgálat és antitestek kimutatása a plazmasejtekben.
A feltételezett T-leukocita módosulásokkal rendelkező beteg teljes körű vizsgálata számítógépes, mágneses rezonancia képalkotást és ultrahangot foglal magában.
A limfóma végső prognózisa átfogó vizsgálaton alapul, és a lézió típusától függ. Az agresszív daganatok azonnali kezelést igényelnek. Ezeknek a daganatoknak a kezelési programja főként kemoterápiát és sugárzást foglal magában. Kielégítő eredmény a sugárterápia utáni pozitív remisszió.
Ez az információ egészségügyi és gyógyszerészeti szakembereknek szól. A betegek ezt az információt nem használhatják orvosi tanácsként vagy ajánlásként.
T-sejtes immunhiányok
Az elsődleges T-sejt-hiányok ritka örökletes rendellenességek, amelyek befolyásolják a T-sejtek fejlődését és működését. Ezek a rendellenességek általában csecsemő- vagy kisgyermekkorban jelentkeznek; azonban a tünetek megjelenésének kora a mögöttes génhibától függően változhat. Bár a T-sejtes immunválaszok szelektíven megzavarhatók, a kóros B-sejt funkciók gyakran társulnak ehhez, részben a kapcsolódó belső B-sejt-defektusok miatt, de azért is, mert a legtöbb antitest-termelés a T-sejt-segítségtől függ.
SÚLYOS KOMBINÁLT IMMUNHIÁNYOS SZINDRÓMA
A súlyos kombinált immunhiányos szindróma (SCID) egy örökletes rendellenesség gyermekeknél, amelyet a T- és B-sejtek mélyen hibás vagy hiányzó funkciói jellemeznek. A SCID gyakran végzetes az élet első évében, a terápiás őssejt-transzplantáció vagy adenozin-deamináz (ADA) hiánya esetén az enzimpótlás ellenére. Az érintett betegek korai azonosítása még az opportunista fertőzések kialakulása előtt kritikus fontosságú a kedvező eredmény eléréséhez. A SCID diagnózisát akkor erősítik meg, ha az érintett gyermek lymphopeniát mutat (A jelenlévő genetikai heterogenitás ellenére az SCID-ben szenvedő betegek élete első 6 hónapjában sok hasonlóságot mutatnak.
Általában az érintett gyermekeknél a következő fertőzések alakulhatnak ki:
Bakteriális
Gram-negatív szepszis
A Bacillus Calmette-Guerin terjesztése immunizálás után
Gombák és protozoonok okozzák
Candidiasis
Aspergillosis
Pneumocystis carinii
Vírusos
Citomegalovírus
Parainfluenza vírusok
Adenovírusok
Légzőszervi syncytialis vírus
Disszeminált varicella (bárányhimlő)
Vakcinával szerzett bénulásos polymyelitis
Molluscum contagiosum
Hasmenés vagy felszívódási zavar miatti súlygyarapodás elmaradása is előfordulhat. Az orvosi kezelésre nem reagáló korai erythemás makulopapuláris kiütés megjelenése anyai T-sejt-transzplantációból eredő krónikus graft-versus-host betegségre (GVHD) utalhat. A legtöbb SCID-ben szenvedő beteg csecsemőmirigy-hipopláziában szenved, és hiányoznak vagy kicsi, fejletlen nyirokcsomók és mandulaszövetek; Hepatosplenomegalia kimutatható az anyai GVHD által érintett gyermekeknél. A mellkasi röntgenfelvételek gyakran hiányzó csecsemőmirigy-árnyékot és enyhe tüdőmintát mutatnak, a jelentős légúti tünetek jelenléte ellenére.
A legtöbb SCID-beteg perifériás CD3+ T-sejtszáma 500 sejt/mm3 vagy kevesebb (normál tartomány 3000-6500 sejt/mm3), valamint változó számú B és természetes gyilkos (NK) limfociták a mögöttes genetikai hibától függően. Az SCID a B- és NK-sejtek jelenléte vagy hiánya szerint T-B+NK+, T-B+ NK-, T-B-NK+, T-B-NK- és atipikus T+B+ szindrómákba sorolható (1. táblázat). Az ADA-hiányban szenvedő betegekben van a legkevesebb a keringő limfociták száma, míg az ismeretlen autoszomális recesszív (AR) T+B+ SCID-ben, ZAP-70-hiányban szenvedő és anyai T-sejtbe jutásban szenvedő betegekben van a legtöbb limfocita, gyakran a normál határokon belül. Az SCID-ben szenvedő betegek reagálnak a késleltetett típusú túlérzékenységi bőrtesztekre, és a T-sejtek működésének in vitro mérései jelentősen, a normál értékek 10%-ára vagy kevesebbre csökkennek.
A keringő T-limfociták kis száma és a súlyos hipogammaglobulinémia mellett az IgG speciális csökkenése is megfigyelhető. A normál határokon belüli szérum IgG-szintek általában az anyai antitesteket tükrözik SCID-ben vagy intravénás gamma-globulinban (IVIG) szenvedő kisgyermekekben. A szérum IgA és IgM szintje a nullától a normál életkorral összefüggő értékekig terjed. A kimutatható szérum IgE és az eozinofília jelenléte általában anyai GVHD-ben vagy Omenn-szindrómában (OS) szenvedő gyermekeknél fordul elő. A szérum immunglobulinok jelenléte ellenére néhány SCID-betegben nem fejlődnek ki antigén-specifikus antitestek; ezért az antitest-függő módszerek, például az enzimhez kötött immunszorbens tesztek alkalmazása a fertőző ágenseknek való kitettség szűrésére SCID-ben szenvedő betegeknél hamis negatív vagy álpozitív eredményeket ad az IVIG beadása miatt. Ehelyett immunfluoreszcenciával vagy láncpolimerizációval történő közvetlen antigéndetektálást kell alkalmazni a fertőzés diagnosztizálására immunhiányos betegeknél.
Az SCID egyetlen kezelése a hematopoietikus őssejtek helyreállítása. Az optimális kezelés a csontvelő-transzplantáció (BMT) vagy a szövetkompatibilis testvérből származó perifériás őssejt-transzplantáció. Sajnos a legtöbb betegnél nincs HLA-azonos családi donor. A szülőktől származó T-sejt-hiányos haptidentikus BMT-t gyakran végeznek, és sok SCID-ben szenvedő betegnél sikeres. A megfelelő, nem rokon BMT vagy köldökzsinórvér őssejt-transzplantációt is alkalmaztak az immunrendszer helyreállítására. A transzplantáció fő szövődményei a graftkilökődés, a GVHD, a fertőzések és a kemoterápia toxicitása. Egyes betegeknek hosszú távú immunszuppresszióra van szükségük a GVHD vagy az élethosszig tartó IVIG szabályozásához, ha a donor B-sejt beültetése sikertelen és a normál B-sejt funkció nem áll helyre.
X-hez kötött súlyos kombinált immunhiányos szindróma
Az X-hez kötött SCID (XSCID) az összes SCID eset 30-40%-át teszi ki, és 100 000 születésből 1-2 esetben fordul elő. Öröklődési módjára számos olyan törzskönyvből következtettek, ahol a következő generációkból származó fiúk kora gyermekkorukban haltak meg kontrollálhatatlan vírusos vagy gombás fertőzések következtében. Az XSCID hibáját 1993-ban az interleukin (IL)-2 receptor közös g-láncaként (gc) azonosították. A későbbi vizsgálatok kimutatták, hogy a gc négy további citokin receptorának is része: IL-4, IL-7, IL-9 és IL-15. Úgy tűnik, hogy a gc molekula nélkülözhetetlen a citokin-citokin receptor kölcsönhatásokból származó aktivációs jelek intracelluláris átviteléhez, amelyek a limfociták proliferációjához és éréséhez szükségesek. A gc-tartalmú receptorkomplexek hiánya a T-sejtek és NK-sejtek fejlődésének korai szuszpenziójában és éretlen B-sejtek termelődésében nyilvánul meg, amelyek az IgG, IgA IgE termeléséhez szükséges hibás izotípusváltást okozzák. Az IL-7 és receptora közötti kölcsönhatás különösen kritikus a limfoid-elkötelezett progenitor sejtek és a pluripotens őssejtek megkülönböztetésében. Úgy tűnik, hogy ez az immunhiány a SCID prototipikus T-B+NK formája.
A legtöbb XSCID által érintett férfibeteg abszolút limfocitaszáma kevesebb, mint 2000 sejt/mm3 a perifériás vérben, kevesebb mint 200 sejt/mm3 CD3+ T-sejttel (tartomány 0-800 sejt/mm3), kevesebb mint 100 sejt/mm3 NK-sejt és a B-limfociták megnövekedett százalékos aránya (gyakran >75%) (2. táblázat). A szérum IgG és IgA szintje rendkívül alacsony, és nincs specifikus antitest termelés. Ezzel szemben a szérum IgM és IgE szintje normálisnak tűnhet az anyai T-sejt beépítés eredményeként. In vitro a T-sejtek és az NK-sejtek funkciói általában gyengék. A legtöbb XSCID-fertőzött gyermek vérében HLA-típusú anyai T-limfociták találhatók. Nyílt GVHD alakulhat ki, ha jelentős számú anyai T-sejt van jelen, amely képes reagálni a szülői úton szerzett HLA-antigénekre. A limfocitózis, a normál IgM- és IgE-szintek, a hepatomegalia, a lymphadenopathia és az anyai GVHD-ből eredő krónikus bőrkiütés gyakran késlelteti az XSCID kimutatását.
Az IL2RG génben több különböző mutációt azonosítottak XSCID utódokban. A cisztás fibrózissal ellentétben az XSCID-ben nem fordul elő általános mutáció; azonban néhány forró pontot azonosítottak egy génben, amelyben gyakran mutációkat azonosítanak. Egyes mutációkról kimutatták, hogy viszonylag csekély hatással vannak a gc működésére; ezeknek a betegeknek általában kisebb a limfopeniája, jobban megőrződött a T-sejt funkciója, és normálisabb a szérum Ig szintje. Ezeket a gyerekeket gyakran úgy osztályozzák, hogy ismeretlen AR T+ B+ SCID-jük van, ami késlelteti a mögöttes genetikai hibájuk felismerését. Mivel a legtöbb XSCID család egyedi mutációkat tartalmaz, az IL2RG gén kódoló régióinak szekvenciális elemzését kell elvégezni a káros mutációk jellemzésére. Anyai DNS-szekvenálás is elvégezhető a mutációk megerősítésére; azonban az XSCID-ben szenvedő férfibetegek több mint 50%-ánál spontán előforduló IL2RG mutáció van, anélkül, hogy a DNS-szekvenálás vagy az X-kromoszóma inaktiváció nem vak mintázata bizonyított volna öröklött anyai X-kromoszómamutációt. A hisztokompatibilis vagy haploidentikus ICH vagy perifériás vagy köldökzsinór-őssejt-transzplantáció biztosítja az immunrendszer helyreállítását a legtöbb XSCID-ben szenvedő fiúban. A méhen belüli VMT sikeresen kezeli az érintett magzatokat, és úgy tűnik, hogy a génterápia valós lehetőséggé válik
JAK3 enzimhiány
A tipikus T-B+NK-SCID fenotípusú lányoknál és néhány fiúnál nincs mutáció az IL2RG génben. Ezeknél a betegeknél, akik közül sokan rokonszülőktől születtek, AR SCID-ben szenvednek, amelyet a JAK3 enzim mindkét alléljának mutációi okoznak. A JAK3 enzimhiányt először 1995-ben írták le, és az összes SCID eset 10-20%-át teheti ki. A JAK3 egy citoplazmatikus tirozin kináz, amely a gc-hez kapcsolódik, és szükséges a citokinekkel kapcsolatos jelek átviteléhez a gc-t tartalmazó citokin receptoroktól. A JAK3 mutációi megzavarják a jelátvitelt ezeken a receptorokon keresztül, ami alátámasztja azt a megfigyelést, hogy a gc működése abszolút függ a downstream JAK3 aktiválódásától. A JAK3 mutációk változóak, és általában minden egyes családban egyediek. Sokan kizárják a JAK3 mRNS vagy fehérje expresszióját, de leírtak kivételeket, amelyek enyhébb SCID fenotípusban és néhány T- és NK-sejtek termelődésében nyilvánulnak meg.
Interleukin-7 receptor hiány
Az IL-7 receptor lánc (IL-7Ra) hibái az AR SCID ritka okai. Az IL-7Ra-hiány fenotípusa jelentős kivételekkel hasonló az XSCID- és JAK3-hiány fenotípusához. Ezekkel az immunhibákkal ellentétben az IL-7Ra-hiány nem akadályozza meg az NK-sejtek fejlődését, és a SCID T-B-NK+ formájának tekinthető.
Interleukin-2 hiány
Egyes családokat publikáltak, amelyekben az IL-2 termelése hibás. Az IL-2-hiányban szenvedő betegek perifériás limfocitaszáma (T+ B+ SCID) és hipogammaalbuminémiája viszonylag normális. In vitro a T-sejtek működése csökken, de rekombináns IL-2 hozzáadásával korrigálható. Ezeknél a betegeknél a mögöttes genetikai hibák nem ismertek, de úgy gondolják, hogy befolyásolják az IL-2 gén transzkripciós szabályozását. A rekombináns IL-2-vel végzett parenterális terápia részleges immunregenerációt biztosíthat ezeknél a betegeknél.
RAG 1 és 2 hiány
A limfocita-specifikus rekombináz-aktiváló gén (RAG) 1-ben vagy 2-ben (RAG1 és RAG2) szenvedő gyermekeket először 1996-ban írtak le. A RAG 1 vagy RAG 2 a T-B-NK+ SCID elsődleges formájaként nyilvánul meg, és az összes eset 10-20%-át teheti ki. A RAG 1 és RAG 2 funkciója nélkülözhetetlen a T-sejt- és B-sejtes antigénreceptorok generációihoz (TCR, illetve BCR). A T-sejt és B-sejt ontogenezis során a TCR immunglobulin gének különböző variábilis (V), divergens (D) és járulékos (J) szegmenseit tartalmazó kromoszómális DNS átrendeződik funkcionális antigénreceptorok előállítására. A V(D)J rekombináció biztosítja az antigénreceptorok sokféleségét és az emberi immunrendszer azon képességét, hogy több mint 108 antigénre reagáljon. A V(D)J rekombináció végrehajtására való képtelenség a T-sejtek és B-sejtek érésének leállásában nyilvánul meg a limfocita differenciálódás korai szakaszában, az összes T- és B-sejt teljes hiányával és az agammaglobulinémiával. Az NK-sejtek nem expresszálnak antigén-specifikus receptorokat, és fejlődésüket nem gátolja a hibás RAG-1 vagy RAG-2 funkció. A T-B-NK+ SCID-ben szenvedő egyedek utódaiban mind a RAG1, mind a RAG2 mutációit azonosították. Súlyos RAG1 mutációk alakulnak ki a belső fehérje fragmentumokban, és gyakoribbak lehetnek, mint a RAG2 rendellenességei.
Omenn-szindróma (OS)
Az OS egy ritka AR-rendellenesség, amelyet Omenn 1965-ben SCID-ként ír le, és a következő tünetek jellemzik:
Fizikai adatok
Erythroderma
Lymphadenopathia
Hepatosplenomegalia
A hasmenés következtében fellépő súlygyarapodás elmulasztása
Generalizált ödéma
Láz
Laboratóriumi adatok
Hipoalbuminémia
Eozinofília (>1000 sejt/mm3)
Változó számú limfociták
Csökkent a CD3+ T-sejtszám növekedéséig
B-sejtek hiánya
Az NK-sejtek normál száma
Egyértelműen hibás a T-sejt- és B-sejt-funkció
Hipogammaglobulinémia
Jelentősen csökkent az IgG, IgM és IgA szintje
Hyper-IgE (>1000 NE/ml)
1998-ban OS-ben szenvedő betegeknél RAG1 vagy RAG2 mutációkat azonosítottak, amelyek részleges V(D)J rekombináz aktivitást és gyengén aktivált, de anergiás, oligo-klonális T-sejtek kialakulását eredményezték.
Ezek a klinikai megnyilvánulások az OS-re jellemzőek, és megkülönböztetik a SCID egyéb formáitól. Sok éven át az OS-t az anyai GVHD súlyos változatának tartották, de a beágyazott anyai T-limfocitákat nem lehetett azonosítani. Az érintett gyermekeknél a perifériás T-limfociták száma jelentősen csökkent a normál szintig. Sok esetben a CD3+ T limfociták száma normális volt, de nem volt keringő B sejt. A szérum immunglobulinok nem voltak kimutathatók, kivéve a jelentősen megemelkedett IgE-szintet (gyakran >1000 NE/ml). A teljes RAG 1 vagy 2 hiányhoz hasonlóan az NK-sejtek száma normális marad az OS-ben szenvedő betegeknél. Az OS-t a SCID T-B-NK+ formájának tekintik, de a ritka sikeres V(D)J rekombinációs események következtében kialakuló oligoklonális T-sejtek jelenléte megzavarja a diagnózist. A hiper-IgE-t a nem limfoid szövetekben ritka B-sejtek jelenléte okozza, amelyeket az IL-4 IL-5 termelésére irányuló helper sejtek IgE termelésére serkentenek, és általában túlérzékenységi vagy allergiás betegségekben fordul elő. A limfadenopátia jelenléte megkülönbözteti az OS-t a SCID más formáitól, amelyekben nincs anyai GVHD. A nyirokcsomók szövettani vizsgálata OS-ben feltárja az építészeti zavarokat, a tüszőképződés hiányát, valamint az eozinofilek, hisztiociták és aktivált T-sejtek túlzott beszűrődését. A bőrt is tömegesen beszivárogtatják a gyulladásos sejtek.
Az OS-ben szenvedő betegek DNS-szekvenálási elemzése olyan mutációkat tár fel a RAG1-ben vagy RAG2-ben, amelyek nem szüntetik meg teljesen a V(D)J rekombináz aktivitást. Ismeretlen okokból a részleges RAG1 vagy RAG2 funkció a TCR produktívabb átrendeződését eredményezi, mint a BCR, így oligoklonális T-sejtek vannak jelen, és a keringő B-sejtek nagyon ritkák. Érdekes módon a legtöbb érintett gyermek nem rokon szülőktől születik. A TDC előtt az anyai GVHD-t ki kell zárni. Ezek a gyerekek gyakran súlyosan megbetegednek lázzal, fehérjevesztő enteropátiával és általános ödémával a bél- és bőrgyulladás miatt. Ablatív kemoterápia és immunszuppresszió szükséges a transzplantátum aktivált autológ T-limfociták általi kilökődésének megakadályozásához. Előnyben részesítik a szövetkompatibilis VMT-t, mivel az OS-ben szenvedő betegeknél nagyobb a haploidentikus transzplantáció sikertelenségének kockázata.
Súlyos kombinált navajo szindróma
A navajo SCID egy AR mutáció, amely az ath-abascan indiánok 2000 élveszületése közül körülbelül 1-ben fordul elő. A mögöttes genetikai hiba továbbra is ismeretlen, de az utódelemzés során a 10p kromoszómához térképezték fel. A navajo SCID klinikai megnyilvánulásai hasonlóak az SCID más formáihoz, kivéve egy szokatlan jelenséget, amelyet a legtöbb betegnél az élet első 4 hónapjában figyeltek meg – a nem herpeszvírussal összefüggő szájüregi és genitális fekélyeket. Az érintett gyermekeknél lymphopenia alakul ki (300-1800 sejt/mm3), a T- és B-sejtszám kevesebb, mint 200 sejt/mm3. In vitro a T-sejtek funkciója és a szérum immunglobulin szintje jelentősen csökken, míg az NK-sejtek bőségesek és normálisak, így a Navajo SCID T-B-NK+ SCID-nek minősül, de nem kapcsolódik hibás RAG-1 vagy RAG-2 funkcióhoz. Az NK aktivitás jelenléte megnehezíti a VMT-t, növelve a transzplantátum kilökődésének kockázatát. Intenzív pre-VMT ablatív kondicionálás szükséges az érintett gyermekek transzplantációjához.
Adenozin-deamináz hiány
Az ADA-hiány az összes SCID-eset körülbelül 15%-át teszi ki. 1972-ben írták le először. A SCID más formáitól eltérően, amelyekben a mutációk specifikusan befolyásolják a T-sejtek és gyakran a B-sejtek működését, az ADA-hiányt az összes sejt metabolikus toxicitása jellemzi, a legkifejezettebb hatással a limfocitákra és a limfoid progenitorokra. Az ADA-hiány a T-B-NK-SCID leggyakoribb formája.
Az ADA egy széles körben elterjedt enzim a purin lebontási folyamatban, amely katalizálja az adenozin és dezoxiadenozin inozinná és dezoxiinozinná történő dezaminációját. Az ADA hiánya ezen szubsztrátok intracelluláris és extracelluláris folyadékokban való felhalmozódásában nyilvánul meg. A limfocitákban a dezoxiadenozin-trifoszfát jelentősen megnövekedett szintje a dezoxiadenozin intracelluláris felvételével és foszforilációjával jár együtt, ami magyarázatot adhat arra, hogy a limfociták miért olyan érzékenyek ezen metabolitok toxikus mellékhatásaira. Az adenozin, dezoxiadenozin és dezoxiadenozin-trifoszfát gátolja a különböző sejtfolyamatokat, beleértve a ribonukleotid-reduktáz aktivitást, ami sejthalálhoz és szövetkárosodáshoz vezet. Az RBC ADA aktivitását jellemzően az ADA-hiány diagnosztizálásának időpontjában mérik, de általában alacsonyabb, mint a limfocitákban.
Az ADA-hiány klinikai és laboratóriumi spektruma meglehetősen széles, és a mögöttes génmutációk súlyosságától függ. Egészséges felnőttek nagy csoportjának szűrése azt mutatta, hogy a normál vörösvértest ADA-aktivitás 7%-a vagy több az ép immunitáshoz kapcsolódik; Ezért az SCID és az ADA-hiány kevésbé súlyos immunhibái az ADA gén mutációihoz kapcsolódnak, amelyek megszüntetik vagy szinte teljesen letiltják az enzim működését. A betegek hozzávetőleg 80%-ánál korán kezdődő, klasszikus ADA-hiány lép fel, és az élet első 3 hónapjában alakul ki. A betegek körülbelül 50%-ánál a megkülönböztető klinikai jellemző a csontrendszeri rendellenességek, a kezdeti köpölyözés, valamint a mellkasröntgenen látható costochondralis csomópontok fokozatos eltűnése vagy kiszélesedése. Ezek a betegek 0,01% vagy kevesebb ADA-aktivitást tartanak fenn, és alymphocytosisban szenvednek (az ADA-hiányos esetek körülbelül 15-20%-át csak 1-2 éves korukban diagnosztizálják; ezek a betegek a kevésbé destruktív hatás miatt késleltetett késői SCID-ben szenvednek. Az ADA gén mutációi Az érintett gyermekek 0,1-2%-os ADA-aktivitást tartanak fenn, a perifériás vér limfocitáinak száma kevesebb, mint 500 sejt/mm3, de kevésbé súlyos hipoalbuminémia az első életévben, azonban a sejtek számának csökkenése. A T- és B-sejtek és funkcióik meglehetősen gyorsan alakulnak ki, és a fertőzés nem sokkal később alakul ki a betegek 5%-ánál vagy annál kevesebbnél, és 3 és 15 éves kor közötti kombinált immunhiányt diagnosztizálnak. tartós herpeszfertőzés és visszatérő felső légúti fertőzések előzték meg, gyakran Streptococcus pneumoniae.
Az autoimmun betegségek, különösen a hemolitikus anémia és a thrombocytopenia későn jelentkező ADA-hiányhoz kapcsolódnak. A laboratóriumi eredmények jellegzetesek, és 2–5%-os ADA-aktivitást, 800 sejt/mm3-nél kisebb keringő limfocitaszámot, 500 sejt/mm3-nél kisebb CD3+ T-sejtszámot és eozinofíliát tartalmaznak. Hipogammaglobulinémia alakulhat ki IgG2 hiányával és az IgE szérumszintjének emelkedésével. In vitro a T-sejtek működése és a specifikus antitest-válaszok, különösen a poliszacharid antigénekre, jelentősen csökkennek. Felnőttkori kezdetű és részleges ADA-hiányos izolált családokat írtak le.
Az eritrociták ADA-aktivitásának mérése nem megbízható transzfúziós betegeknél; ehelyett a limfocitákban vagy fibroblasztokban mérni kell az enzimműködést az ADA-hiány diagnózisának megerősítésére. Az ADA-hiány prenatális diagnózisát a magzatból nyert sejtek enzimaktivitásának szűrésével végzik, de a megerősítéshez DNS-szekvenálásnak kell kísérnie. Több mint 60 különböző mutációt azonosítottak immunhiányos betegekben. Legtöbbjük olyan DNS-szekvenciákba csoportosul, amelyek a szubsztrátkötésben részt vevő vagy katalitikus funkciójú aminosavakat kódolnak. Az ADA-hiányban szenvedő betegek mutációinak több mint 50%-a teljesen letiltja az enzimaktivitást, és korai SCID-t eredményez. Az AR SCID más formáitól eltérően, amelyekben a betegek hajlamosak homozigóták lenni ugyanarra a mutációra, a legtöbb ADA-hiányos beteg kevert heterozigóta két különböző mutáns ADA allél tekintetében.
Az ADA-hiányos SCID optimális terápiája a szövethez illesztett BMT. Úgy tűnik, hogy a haploidentikus BMT beültetése még a transzplantáció előtti abláció mellett is csökken ADA-hiány esetén más SCID-diagnózisokhoz képest. A halálozási arány is növekedhet. Ezen okok miatt haploidentikus BMT-t nem végeznek rutinszerűen az ADA-hiány kezelésére; ehelyett az érintett betegeket potenciálisan megfelelő szöveti donor hiányában gyakran ADA helyettesítő terápiával kezelik. Az enzimpótlás nem igényli az ADA limfocitákba való felvételét ahhoz, hogy jótékony hatást gyakoroljon a T- és B-sejtek működésére. Toxikus metabolitok vannak jelen az extracelluláris folyadékban, és egyensúlyban vannak az intracelluláris termékekkel; ezért a metabolitok plazmaszintjének csökkenése a parenterálisan beadott ADA általi lebomlása révén az intracelluláris metabolitok koncentrációjának csökkenését eredményezi. A polietilén-glükollal kötött (PEG) marhahús ADA-t 1986 óta használják, és az érintett betegek immunrendszerének részleges helyreállítását, valamint hosszú távú javulást és túlélést biztosított. A korai SCID-ben szenvedő betegek jellemzően 30-60 E/kg/hét PEG-ADA-t kapnak; az adag csökkenthető gyermekeknél az életkor előrehaladtával. A PEG-ADA terápia hátrányai közé tartozik a magas költség, valamint a gyakori adagolás és a metabolitszintek szoros monitorozásának szükségessége.
A PEG-ADA-t kapó gyermekek immunrendszerének helyreállítása nem teljes, a B-sejtek száma és funkciója jobb, mint a T-sejteké. A betegek továbbra is lymphopéniások maradnak (az ADA-hiány volt az első olyan rendellenesség, amelyet hematopoietikus sejt génterápiával kezeltek. Néhány ADA-hiányos beteg, köztük három újszülött, ADA-gén korrigált autológ érett T-limfocitákat vagy csontvelői vagy köldökzsinór-őssejteket kapott. Bár úgy tűnt, hogy az ADA-t A korrigált limfociták szelektív növekedési előnnyel rendelkeznek az ADA-hiányos sejtekkel szemben, a betegek nem gyógyultak meg a PEG-ADA-kezeléstől, és nem kaptak teljesen PEG-ADA-t ADA-hiány kezelésére.
ATIPIKUS SÚLYOS KOMBINÁLT IMMUNHIÁNYOSSÁG ÉS KOMBINÁLT IMMUNHIÁNYOSSÁG
ZAP hiány -70
A ZAP-70 hiány egy ritka AR SCID, amelyet először 1994-ben írtak le izolált rokon családok körében. A limfocitózis (4000-20 000 sejt/mm 3), a túlzott (55-75%) CD 3+ CD 4+ és kevesebb, mint 5% CD 8+ jellemző a T+B+ SCID ezen új formájára. A legtöbb érintett beteg csökkent szérum IgG-szintet, károsodott B-sejtes válaszokat és a T-sejt-funkció hiányát mutat, beleértve az allogén bőrtranszplantátumok gyermek általi elutasítását.
A CD 8+ T-sejtek hiánya a protein tirozin-kináz, a ZAP-70 mutációihoz kapcsolódik. A ZAP-70 funkció kritikus fontosságú a TCR-ből származó jel továbbításában, és ennek hiánya abnormális timociták differenciálódását és hibás T-sejt-aktivációt eredményez, amely szükséges a T-sejtek proliferációjához és működéséhez. Eddig kevesebb, mint 15 ZAP-hiányos beteget írtak le, többségükben kis részükben mutáció van. ZAP-70 a fehérje stabilitását és katalitikus működését befolyásoló gének. DNS-szekvenálás szükséges a két mutáns öröklődésének megerősítéséhez ZAP-70 allélek SCID-betegekben, amelyet a keringő CD 8 + T-sejtek hiánya jellemez.
Hiányosságok a CD 3
A TCR-ek a CD 3-mal kapcsolatban állnak össze és expresszálódnak a felületen, amely hat alegység komplexe (pl. ed és xx). A CD 3 hiány két formáját írták le külön családokban, amelyekben ezeket a CD 3 fehérje mutációi okozzák, ami hibás TCR expressziót eredményez. Bár nem olyan súlyos, mint a rekombinációs hibák, a CD 3g és CD 3e mutációi enyhe vagy közepes immunhiányt okoznak. Az érintett betegek T-sejtszáma és funkciója csökkent; A B-sejtek különböző mértékben érintettek.
Tiszta limfocita szindrómák
A tiszta limfocitás szindrómák (BLS) a szelektív T-sejtes immunhiányokhoz hasonlítanak, de egyes betegekben nem különböztethetők meg a SCID-től. A BLS két típusa a fő hisztokompatibilitási komplex (MHC) osztályú (HLA A, BC) vagy II. osztályú (HLA DR, DQ vagy DP) molekulák hematopoietikus sejteken való hiányos expresszióját tükrözi. Az 1BLS vagy MHC I. osztályú hiány diagnózisa akkor javasolt, ha a limfocitákon lévő HLA A, B és C molekulák szerológiai módszerekkel nem mutathatók ki. Ezt a rendellenességet kis számú rokon családban azonosították, és klinikai megjelenése változó. A 2-es típusú BLS-sel ellentétben a legtöbb 1-es típusú BLS-ben szenvedő beteg gyermekkorában tünetmentes; bár ezeknél a gyermekeknél az első tíz életévben tartós légúti fertőzések és krónikus tüdőbetegségek alakulnak ki. A diagnózis késik, mert a betegek normális számú perifériás T- és B-sejttel rendelkeznek, és viszonylag megőrzött immunfunkciókkal rendelkeznek. A limfocita fenotípus kimutathatja a CD 8+T sejtek csökkenését.
Az 1-es típusú BLS számos specifikus gén egyikében mutatkozik meg. Egyes betegeknél az MHC 1. osztályú gének transzkripciója hibás, míg másoknak mutációi vannak a transzporter antigén-feldolgozáshoz kapcsolódó (TAP)-1 vagy TAP-2 génekben. Ezek a fehérjék részt vesznek a feldolgozott antigének szállításában a citoplazmából az endoplazmatikus retikulumba. TAP-1 vagy TAP-2 hiányában az MHC I. osztályú molekulák az endoplazmatikus retikulumban nem tudnak antigénekkel feltöltődni, ami degradációjukban és csökkent sejtfelszíni expressziójukban nyilvánul meg. Az 1-es típusú BLS kezelése szupportív, és a VMT általában nem javallt.
A 2-es típusú BLS vagy MHC II. osztályú hiány az AR T+B+ SCID meglehetősen ritka formája, amely elsősorban az észak-afrikai vagy mediterrán eredetű rokon családokban született gyermekeket érinti. A II. osztályú MHC-hiányban szenvedő betegeknél gyakran alakul ki krónikus Cryptosporoium fertőzéssel összefüggő májbetegség. A betegekben normális számú keringő limfociták találhatók, CD 4 + T-sejtek számának csökkenése, hipogammaglobulinémia és jelentősen hiányos in vitro T-sejt- és B-sejt-proliferáció specifikus antigénekhez.
A 2-es típusú BLS egyik jellemzője a limfociták csökkent képessége az allogén limfociták stimulálására in vitro tenyészetekben. Ezt a megfigyelést alátámasztja az a megállapítás, hogy az MHC II. osztályú molekulák a normál intenzitás 5%-ánál kisebb mértékben expresszálódnak az érintett betegek vérképző sejtjein; azonban a DNS szekvenálási analízis nem mutatott ki mutációkat az MHC II. osztályú génekben; inkább a betegek öröklött mutációi vannak a transzkripciójukban fontos gének egyikében, különösen a CIITA-ban, az RFX-5-ben, az RFX-B-ben vagy az RFX-AP-ben. A 2-es típusú BLS-ben szenvedő betegek hosszú távú prognózisa katasztrofális. A legtöbb beteg progresszív szervi elégtelenségben hal meg. A szövetkompatibilis vagy haploidentikus VMT általában nem biztosítja az immunrendszer helyreállítását és a túlélés meghosszabbítását a felnőttkorba való átmenet során.
Purin nukleozid foszforiláz hiány
A purin nukleozid foszforiláz (PNP) hiánya egy ritka AR kombinált immunhiány, amely immunhiányokkal és neurológiai tünetekkel jár, beleértve a jelentős fejlődési késéseket, viselkedési problémákat és motoros rendellenességeket. A neurológiai deficittel járó SCID-t PNP-hiányosnak kell tekinteni, amíg az ellenkezőjét be nem bizonyítják. A PNP-hiány klinikai megnyilvánulásai lényegében megegyeznek a SCID-vel; a PNP-t azonban gyakran a kombinált immunhiányos rendellenességek közé sorolják, mivel a B-sejt-hibák viszonylag enyhék korai gyermekkorban. Jellemzően a betegek lymphopeniában szenvednek (in vitro T-sejt funkció. A B-sejtek számának és működésének csökkenése idővel jelentkezik, ami a szérum IgG és IgA alacsony szintjében nyilvánul meg. A B-sejt funkció jelentős csökkenése ellenére több mint 30%-ban betegeknél autoimmun betegségek alakulnak ki, mint például a hemolitikus vérszegénység, a vérlemezke purpura és a vasculitis.
A PNP kíséri az ADA-t, és megelőzi a hipoxantin-foszforibozil-transzferázt a purin lebontási útvonalon. Az inozin-dezoxiinozin és guanozin-dezoxiguanozin hipoxantinná, illetve guaninná történő foszforilációját katalizálja. A guanozin és a dezoxiguanozin toxikusnak tűnik a T-limfocitákra, és az intracelluláris guanozin-trifoszfát (GTP) csökkent szintje elsősorban a központi idegrendszert károsíthatja. A PNP-funkció hiányát jellemzően 1 mg/dl-nél kisebb szérum húgysavszint mutatja, amely felhasználható ennek a rendellenességnek a szűrésére. Az eritrocita PNP aktivitás mérése a diagnózis megerősítésére szolgál. A DNS-szekvenálás feltárta az egyedi mutációk öröklődését a legtöbb családban. Sok PNP-hiányban szenvedő beteg rokon szülőktől született, és olyan mutációik vannak, amelyek teljesen megszüntetik a fehérjeexpressziót és az enzimaktivitást. A PNP-hiány kilátásai rendkívül rosszak. Csak néhány beteg fejezte be sikeresen a vérképzőszervi őssejt-transzplantációt BMT céljából. A GVHD jelentős szövődmény, és nem javulnak a neurológiai tünetek a VMT után. A PNP-hiányra jelenleg nem áll rendelkezésre enzimpótló és génterápia.
Ataxia telangiectasia és változatai
Az ataxia telangiectasia (AT) egy összetett AR-rendellenesség, amelyet agyi ataxia, oculocutan telangiectasia, celluláris sugárérzékenység, rosszindulatú daganatokra való hajlam és kombinált immunhiány jellemez. Az AT világméretű prevalenciája 10 6 élveszületésből 3. Az AT mutált génterméket (ATM) 1995-ben azonosították, és egy fehérje jelátviteli kináz, amely részt vesz a sejtciklus szabályozásában, a DNS-rekombinációban, az apoptózisban és a DNS-károsodásra adott egyéb sejtválaszokban.
Az AT fő tünete a neurológiai károsodás, amely általában ataxiás járásként nyilvánul meg a második életévben, de 4 éves korig a betegek több mint 84%-ánál figyelhető meg. Az ataxia fokozatosan érinti a törzset, a végtagokat és a palatális izmokat, ami dysarthriás beszédben, nyáladzásban, szem apraxiában, koreoathetózisban és a 10. életévben a szabad mozgás képtelenségében nyilvánul meg. A sclera és a bőr telangiectasia ezt követően 4 és 8 éves kor között alakul ki. Az AT sejtes és humorális immunitása változó, de gyakran visszatérő felső és alsó légúti fertőzésekhez és krónikus tüdőbetegségekhez vezet. A legtöbb 3 és 6 év közötti AT-betegnél a bakteriális és vírusos fertőzések megnövekedett előfordulási gyakorisága figyelhető meg, és az IVIG-terápia hatására enyhén csökken. Az AT fontos jellemzője a T-sejtes és B-sejtes neoplazmákra, különösen a leukémiára, valamint a Hodgkin- és non-Hodgkin limfómákra való kifejezett hajlam. A rosszindulatú daganatok a második leggyakoribb halálok az AT-s betegek körében, csak a tüdőbetegségeket és az aspirációs tüdőgyulladást előzik meg.
A genetikai instabilitás, amely kromoszómális transzlokációkban és túlzott DNS-törésekben nyilvánul meg, az AT alapvető jellemzője, és rákot és immunhiányt okozhat az érintett gyermekekben. Az AT további klinikai megnyilvánulásai a növekedési retardáció, a hipogonadizmus és a pubertás késői kezdete, valamint az inzulinrezisztens, nem ketonikus diabetes mellitus. Az a-fetoprotein (AFP) és a karcinoembrióta antigén emelkedett szérumszintje meglehetősen gyakori, és hasznos lehet a diagnózis felállításában.
Az AT-betegek progresszív limfopéniában szenvednek, amely mind a T-, mind a B-sejteket érinti, beleértve a CD 4 + T-sejtek szelektív elvesztését a normál CD 4 - CD 8 arány megfordításával összefüggésbe hozható a túlzott DNS-törések kialakulásával a TCR-ben és az immunglobulin génben a 7., illetve a 14. kromoszómán. Az AT-betegek csökkentett in vitro T-sejt-proliferációs választ mutatnak, de az immunrendszeri diszfunkció súlyossága változó. A humorális hibák közé tartozik az IgA és IgE hiánya a legtöbb AT-betegnél, valamint az izohemagglutinin és a szérum IgG 2 szintje csökkenése az egyének kisebb részében. A specifikus antitest válaszok is károsodhatnak.
Az ionizáló sugárzással szembeni fokozott érzékenység és a genetikai instabilitás meglehetősen gyakori az AT-ben szenvedő betegek körében. Az ATM aktiválhatja a sejtciklus mérföldköveit, válaszul a DNS-károsodásra, beleértve a normál V )D )J rekombinációs eseményeket a T- és B-sejtekben. Az ATM által közvetített sejtciklus-szabályozás rendellenességei nem akadályozzák meg a DNS-szintézis folytatását a DNS-károsodás ellenére, amit a kromoszómatörmelékek idővel felhalmozódó felhalmozódása és a sejthalál bizonyít. A timociták, az éretlen B-limfociták, valamint a központi idegrendszer sejtjei, valamint a vaszkuláris endotélium érzékenyebbek lehetnek ezekre a jelenségekre.
A legtöbb AT-beteg heterozigóta két különböző ATM-mutáció miatt, amelyek befolyásolják a fehérje stabilitását. Sajnos a celluláris sugárérzékenység nem teszi lehetővé a transzplantációt, mint életképes kezelési lehetőséget az AT számára. Ezenkívül a standard kemoterápiás protokollok nem használhatók az AT-vel összefüggő rosszindulatú daganatok kezelésére. Egyes AT-betegek több évig is túlélnek limfoid daganatokkal, de a hosszú távú prognózis katasztrofális. A legtöbb AT-beteg nem éli túl élete harmadik évtizedét. Az AT heterozigóta egyedek is hajlamosak lehetnek a daganatokra.
Az AT variánsok közé tartozik a Nijmegen gap syndroma (NBS), amelyet szintén T-sejtes és B-sejtes immunhibák, sugárérzékenység, genetikai instabilitás és rákra való hajlam jellemez. Az NBS abban különbözik az AT-től, hogy az érintett gyermekek több mint 75%-ánál mikrokefália van jelen, enyhe mentális retardáció és arcdiszmorfizmus, valamint nincs telangiectasia, progresszív ataxia és emelkedett AFP-szint. Az NBS-ben szenvedő betegek mutáns génje a Nibrint kódolja, egy ATM-szerű fehérjét, amely részt vesz a DNS-javításban és a sejtciklus szabályozásában. Az NBS-ben szenvedő betegek immunhiánya meglehetősen jelentős, és magában foglalja a limfopéniát, a CD 4 + sejtszám csökkenését és a CD 4-CD 8 arány inverzióját, a megnövekedett NK sejtszámot és a csökkent in vitro T-sejt funkciót. Alacsony vagy hiányzó szérum IgG és IgA normál vagy emelkedett IgM-szinttel a betegek több mint 90%-ában fordul elő. IgG 2 hiány és a pneumococcus antitestek hibás termelése is kialakulhat.
Wiskott-Aldrich szindróma
A Wiskott-Aldrich-szindróma (WAS) egy X-hez kötött örökletes immunhiányos rendellenesség, amelyet ekcéma, kis vérlemezkékkel járó veleszületett thrombocytopenia és visszatérő fertőzések jellemeznek. 106 gyermekből körülbelül 1-2 gyermeket érint. A WAS gén termékét WAS proteinként (WASP) azonosították 1994-ben. Később kimutatták, hogy az örökletes X-hez kötött thrombocytopenia (XLT) a WASP mutációinak eredménye. A WASP egy intracelluláris jelátviteli molekula, amely más fehérjékkel társulva részt vesz a TCR jel vezetésében, fontos szerepet játszik a citoszkeletális szervezet működésének szabályozásában.
A genotípus és a fenotípus között szoros összefüggés van a WAS-ban. Bár minden betegnek vannak vérlemezke-rendellenességei, a legsúlyosabb WAS-mutációval rendelkező férfiakat (az esetek körülbelül 30%-át teszik ki) a legnagyobb a vérzés, fertőzés, autoimmunitás vagy rosszindulatú daganat okozta halálozás kockázata. Az XLT-ben vagy enyhe WAS-ban szenvedő betegeknél előfordulhat ekcéma és különböző immunhiányos állapotok, de nem alakul ki rák vagy autoimmun betegség. A súlyos vagy klasszikus WAS-ban szenvedő betegek újszülöttkori vagy korai gyermekkorban gyakran jelen vannak petechiákkal és thrombocytopenia okozta vérzéssel (általában 10 000-50 000 sejt/mm3). A véres hasmenés, a koponyaűri vérzés és a köldökzsinór-maradványból vagy körülmetélés utáni túlzott vérzés néha a klasszikus WAS kezdeti megnyilvánulása. A diagnózis a vérlemezkék méretének mérésével igazolható. Az idiopátiás thrombocytopeniás purpurával ellentétben a vérlemezke térfogata WAS-ban átlagosan 3,8-5,0 fL (normál tartomány: 7,1-10,5 fL). Bár a WAS nem reagál szteroidokra vagy IVIG-re, súlyos thrombocytopenia esetén splenectomia elvégezhető, de növeli a szepszissel összefüggő halálozás kockázatát; azonban ezt követően az akut idiopátiás thrombocytopeniás purpurához hasonló krízisek lépnek fel, amelyek életveszélyes vérzéssel járhatnak. A WAS-ban bekövetkezett nem BMT-halálozások körülbelül 25%-át vérzéses szövődmények okozzák.
Az ekcéma a betegek több mint 80%-ában alakul ki, és gyakran 6 hónapos életkor előtt jelentkezik. Klasszikus WAS-ban szenvedő fiúknál az első 2 életévben visszatérő szinopulmonális fertőzések alakulnak ki kapszulázott mikroorganizmusokkal. Kevésbé súlyos fertőzések észlelhetők XLT-ben vagy gyenge WAS-ban szenvedő betegeknél. A T-sejtek működésének csökkenésével az opportunista fertőzések, például a tüdőgyulladás által okozott Pneumocystis cariniiés a herpeszvírus által okozott visszatérő fertőzések. A WAS-ban bekövetkezett nem BMT-halálozások körülbelül 40-50%-át fertőzések okozzák.
Azoknál a klasszikus WAS-ban szenvedő betegeknél, akik nem estek át transzplantáción, a leukémia, a hasi és központi idegrendszeri limfómák, valamint az Ebstein-Barr vírussal (EBV) összefüggő daganatok gyakorisága jelentősen megnőtt, ami az összes nem BMT-halálozás 25%-át teszi ki. Autoimmun betegségek, beleértve a hemolitikus anémiát, ízületi gyulladást, vasculitist, gyulladásos bélbetegséget és glomerulonephritist, a súlyos WASP-mutációval rendelkező, átültetett betegek körülbelül 40%-ánál figyelhetők meg, és növelhetik a későbbi rosszindulatú daganatok kockázatát. Rosszindulatú daganatok és autoimmunitás nem fordul elő XLT-ben vagy gyenge WAS-ban szenvedő betegeknél.
Az érintett betegekben a perifériás vér limfocitáinak normál számát gyermekkorban észlelik. Lymphopenia (CD3 + és CD 8 + T-sejtek általában 6 éves korban figyelhetők meg. Ezzel szemben a B-sejtek és NK-sejtek száma normális marad. A legtöbb betegben károsodott a T-sejtek működése. Normál szérum IgG-szint és csökkent IgM-szint A pneumococcus antitestek hibás termelődése és az izohemagglutinin hiánya következetesen megfigyelhető a WAS-ban szenvedő betegeknél. Mivel a WAS-betegek károsodott T- és B-sejt-funkciókat mutatnak, legtöbbjük IVIG-t kap.
A hematopoietikus őssejt-transzplantáció az egyetlen kezelési lehetőség a klasszikus WAS számára. Az immunrendszer és a vérlemezke helyreállításának legnagyobb sikere azoknál a gyermekeknél érhető el, akik 5 éves koruk előtt szövetkompatibilis BMT-t kaptak. A köldökzsinórvér őssejt-transzplantáció és a hozzájuk illő, nem rokon BMT szintén sikeresnek bizonyult kisgyermekeknél. A transzplantáció előtti abláció szükséges a donor T-sejtek és vérlemezkék beépülésének biztosításához. A haploidentikus BMT-ben a graftkilökődés riasztóan magas, de a sikeres transzplantáción átesett WAS-betegek meggyógyulnak az immunhiányból és a betegség egyéb szövődményeiből. A WASP-ben több mint 100 mutációt írtak le, és ezek közül sok egyedi az érintett családokra, azonban a WAS gén néhány hotspotját azonosították, ahol a mutációk a legmagasabb gyakorisággal fordulnak elő. A klasszikus WAS-t általában olyan mutációk okozzák, amelyek elnyomják vagy megszüntetik a fehérjeexpressziót.
X-hez kötött limfoproliferatív rendellenesség
Az X-hez kötött limfoproliferatív (XLP) betegség vagy Duncan-szindróma egy kombinált immunhiány, amely az Ebstein-Barr vírusok (EBV) expozícióját követően jelentkezik. Az XLP gyakorisága körülbelül 1-3 10 6 fiúból. Az XLP (LYP) génről 1998-ban kimutatták, hogy a T-sejt-specifikus fehérjét, az SAP-t (signaling lymphocyte activation molekula-asszociált fehérjét) kódolja. Az SAP fontos szerepet játszik az aktivált T-sejtek jelátvitelének szabályozásában, és az SAP-hiányos T-sejtek nem tudják szabályozni az EBV által kiváltott B-sejt-proliferációt; ezért az immunrendszer csökkent képessége az EBV-vel fertőzött B-limfociták eltávolítására XLP-ben inkább az EBV-specifikus T-sejtes válaszok hibáival jár, mint a B-sejtekben.
Az EBV-fertőzés előtt a legtöbb XLP-mutációban szenvedő fiú klinikailag egészséges. Az EBV-nek való kitettség az esetek 60%-ában fulmináns és gyakran végzetes fertőző mononukleózist okoz. Súlyos hepatitis, vírussal összefüggő hematophagocytás szindróma, aplasztikus anaemia és többrendszerű szervi elégtelenség, amelyet halállal kísér, az esetek 95%-ában a fertőző mononukleózis megjelenését követő 8 héten belül alakul ki. A hasi és központi idegrendszeri Hodgkin és non-Hodgkin limfómák az XLP-ben szenvedő fiúk körülbelül 30%-ánál alakulnak ki. Nagyon kevés EBV-vel összefüggő limfoproliferatív betegségben szenvedő beteg él 10 évnél tovább. Az emelkedett szérum IgM-szinttel járó hypogammaglobulinémiában szenvedő betegek prognózisa kedvező, hacsak nem társul limfoproliferatív betegséggel. Az XLP-s betegek prognózisa katasztrofális, a legtöbb gyermek gyermekkorban vagy serdülőkorban meghal. A vírusellenes vagy immunszuppresszív gyógyszerek nem hatékonyak a fulmináns betegségek kezelésében. Csak kisszámú betegnél tapasztalták az immunrendszer helyreállását a transzplantáció után; a transzplantáció azonban összefügg a beteg életkorával: a legtöbb 15 év feletti beteg nem éli túl a BMT-t.
Az EBV fertőzést követően az XLP-betegek T-sejt- és B-sejt-hibákat mutatnak. Bár a betegekben normális a T-sejtek száma, a CD 8 + T sejtek dominálnak, és a normál CD 4 - CD 8 arány megfordul. Az EBV-asszociált nukleáris antigén elleni antitesttiterek sok esetben hiányoznak vagy jelentősen csökkentek, míg a kapszid antigén elleni antitest-termelés változó. Az NK-funkció rendellenességei az érintett fiúk 30%-ánál voltak nyilvánvalóak.
SZELEKTÍV T-SEJT HIBA
DiGeorge szindróma
A komplex veleszületett szív- és arckoponya-rendellenességekben szenvedő gyermekek több mint 90%-ánál, amelyekről kezdetben különböző genetikai szindrómákat, köztük DiGeorge-szindrómákat (DGS), Shprintzen-kórt, velokardiális és conotruncalis arcrendellenességeket diagnosztizáltak, kimutatták, hogy egy példányban gyakori mikrodeléció. A hemizigóta kromoszóma 22q 11.2 mikrodelécióról azt gondolják, hogy az érintett gyermekek számos egészségügyi problémájával, köztük a fejlődési késleltetéssel és az immunhiányos állapottal járnak összefüggésbe. Fluoreszcenciás in situ hibridizációs (FISH) analízissel ez a kromoszóma-rendellenesség bármely gyermeknél és annak korábban fel nem ismert családtagjainál diagnosztizálható.
A DGS klasszikusan magában foglalja a conotruncalis szívhibákat, amelyek perzisztáló hipokalcémiával és celluláris immundeficienciával társulnak, másodlagosan a harmadik és negyedik garattasak területének kiterjedtebb fejlődési rendellenességei miatt, amelyek a mellékpajzsmirigyet és a csecsemőmirigyet érintik. A DGS-hez leggyakrabban kapcsolódó specifikus szívelégtelenségek a következők: az aortaív megszakadása, fallot tetralgia és truncus arteriosus. A DGS és más 22q 11.2 kromoszóma deléciós rendellenességek fenotípusos expressziója még családon belül is változik, és gyakran szájpadhasadékot és arcrendellenességeket is magában foglal.
A 22q 11.2 kromoszóma FISH által kimutatott mikrodeléciói eltérő méretűek, de általában az úgynevezett DiGeorge-kritikus régióhoz vagy DGS -1 lókuszhoz kapcsolódó szekvenciát tartalmaznak. Az esetek körülbelül 25%-ában a deléció örökletes, gyakran fenotípusosan egészséges szülőtől származik. A 22q 11.2 kromoszóma fennmaradó deléciói de novo fordulnak elő az érintett gyermekekben. Ennek a kromoszóma-rendellenességnek a prevalenciája 100 000 élveszületésre számítva legalább 13, így ez a veleszületett szívbetegség leggyakoribb genetikai oka; azonban a 22q 11,2 deléciós szindrómában szenvedő gyermekek 20%-ának nincs szívelégtelensége. A részletes térképezési elemzés lehetővé tette egy minimális DiGeorge-kritikus régió azonosítását, de a 22q 11.2 kromoszóma deléciós szindrómáért felelős specifikus gén(eke)t nem találtuk meg. Ennek ellenére ezek a rendellenességek a neurális keresztmigrációban részt vevő gének hoploinsufficienciájából erednek. Az biztos, hogy mivel a 22q 11.2 deléciós szindrómában szenvedő gyermekek és az érintett családtagjaik fenotípusai sokféle fizikai leletet tartalmaznak, több gén és módosító tényező határozza meg azok okát. Ezen túlmenően, a DGS fenotípusos megnyilvánulásait mutató ritka betegeknél nem észlelhető a 22q 11.2 kromoszóma mikrodeléciója, ehelyett néhány beteg homozigóta a DGS-II lókuszt vagy lókuszt meghatározó 10p kromoszóma deléciója miatt.
A DGS immunhibáinak spektruma meglehetősen széles, de leggyakrabban a CD3+ T-limfociták számának csökkenését, 1000 sejt/mm 3 alatti CD4+ T-sejtszámot és enyhén károsodott sejtes immunitást foglal magában A normál T-sejtek 50%-a vagy kevesebb, az in vitro T-sejt-proliferációs válasz pedig kevesebb, mint a normál 50%-a Egyes immunológusok úgy vélik, hogy a DGS diagnózisát azokra a gyermekekre kell korlátozni, akiknél kevesebb a kromoszómális 22q 11.2 deléciós szindróma. mint 500 CD 3+ T-sejt/mm 3 A kutatók általában úgy vélik, hogy a T-sejtek érése normális a DGS-ben szenvedő betegeknél, és a limfopenia közvetlenül összefügg a funkcionális csecsemőmirigy tömegének mennyiségi csökkenésével. Nincsenek laboratóriumi vizsgálatok vagy fenotípusos markerek. immunológiai kimenetel Az immunhiba általában javul az életkorral, és sok gyermek normális számú funkcionális T-limfocitát termel a második életév végére. azonban az immunrendszer helyreállításának sebessége és mértéke nem megjósolható, és a DGS-ben szenvedő betegek körülbelül 5%-ánál jelentősen csökkent a T-sejtszám és -funkció a thymus aplasia következtében; ezeknek a betegeknek szüksége lehet BMT-re. A betegek 50 százalékának humorális immunitása is károsodott. A DGS-ben szenvedő gyermekek immunhiányának következményei közé tartozik a vírusfertőzésekkel szembeni fokozott fogékonyság. Ez a megállapítás az élő vírusokkal történő immunizálás elhalasztásával a gyakorlatban is érvényesül addig, amíg az immunkompetencia bizonyítéka nem áll rendelkezésre. A szokásos kritérium a normál T-sejtek száma, a normál in vitro T-sejt-proliferációs válaszok legalább 75%-a, valamint a tetanusz-toxoid immunizálást követő specifikus antitest-termelés. A 22q 11.2 kromoszóma deléciós szindrómában szenvedő gyermekek immunhibáinak jelenléte és súlyossága változó, és nem korrelál más fenotípusos megnyilvánulásokkal. Ezen túlmenően, a T-sejtes immunhiány olyan gyakran alakul ki a nem DGS kromoszómális 22q 11.2 deléciós szindrómával diagnosztizált gyermekeknél, mint a klasszikus DGS-ben szenvedő gyermekeknél.
Egészen a közelmúltig sok 22q 11.2 kromoszómális deléciós szindrómában szenvedő gyermek halt meg szívhibája következtében. Ezek a gyerekek túlélik a felnőttkort, és krónikus betegségeik új aspektusai jelennek meg. A kromoszómális 22q 11.2 deléciós szindróma felismert megnyilvánulásai jelenleg az autoimmunitás, a jelentős tanulási nehézségek, a viselkedési és érzelmi diszfunkciók, valamint a pszichiátriai diagnózisok. Fontos, hogy családi konzultációt folytassunk a 22q 11.2 kromoszómális deléciós szindróma ezen megnyilvánulásairól.
Krónikus mucocutan candidiasis
A krónikus mucocutan candidiasis (CMC) a betegségek heterozigóta csoportja, amelyet a körmök, a bőr és a nyálkahártyák tartós gombás fertőzése jellemez. A betegek több mint 50%-ának poliendokrinopátiával jellemezhető AR-betegsége van, és mutációi vannak a transzkripciót szabályozó fehérjét kódoló AIRE génben. Az izolált SMS-ben szenvedő gyermekek mögöttes genetikai hiba nem ismert. A legtöbb beteg limfocitaszáma és funkciója normális; néhány izolált SMS-ben szenvedő betegnél azonban megkülönböztető eredmény az, hogy in vitro T-sejt-proliferáció hiányzik a candida antigénhez.
ÖSSZEFOGLALÁS
A gyermekkorban diagnosztizált örökletes immunhiányok többségét a T-sejtes immunhiányok teszik ki. A legtöbb sejtes immunhiány különböző klinikai és laboratóriumi megnyilvánulások humorális hibáihoz kapcsolódik. A mögöttes genetikai hibák ismertek és meghatározottak a legtöbb öröklött T-sejtes immunhiba esetén, és a mutációelemzés egyre inkább az értékelés és a diagnózis szerves részévé válik. A családdal folytatott részletes megbeszélés a fenotípus-genotípus összefüggésről kritikus fontosságú az orvosi kezelés és a hosszú távú prognózis szempontjából.
Forrás : Melisa E. Elder, / T-CELL IMMUNDEFICIÁK / ÉSZAK-AMERIKAI GYERMEKklinikák 47. évf. N 6.