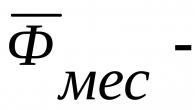Od roditelja dijete može steći ne samo određenu boju očiju, visinu ili oblik lica, već i naslijeđene. Šta su oni? Kako ih možete otkriti? Koja klasifikacija postoji?
Mehanizmi nasljeđa
Prije nego što govorimo o bolestima, vrijedi razumjeti koje su sve informacije o nama sadržane u molekuli DNK, koja se sastoji od nezamislivo dugog lanca aminokiselina. Izmjena ovih aminokiselina je jedinstvena.
Fragmenti lanca DNK nazivaju se geni. Svaki gen sadrži integralnu informaciju o jednoj ili više osobina organizma, koja se prenosi sa roditelja na djecu, na primjer, boja kože, kose, karakterne osobine itd. Kada su oštećeni ili im je poremećen rad, genetske bolesti koje se nasljeđuju pojaviti.
DNK je organizirana u 46 hromozoma ili 23 para, od kojih je jedan polni hromozom. Kromosomi su odgovorni za aktivnost gena, kopiranje i oporavak od oštećenja. Kao rezultat oplodnje, svaki par ima jedan hromozom od oca, a drugi od majke.
U ovom slučaju, jedan od gena će biti dominantan, a drugi će biti recesivan ili potisnut. Pojednostavljeno rečeno, ako se pokaže da je očev gen odgovoran za boju očiju dominantan, tada će dijete tu osobinu naslijediti od njega, a ne od majke.
Genetske bolesti
Nasljedne bolesti nastaju kada dođe do poremećaja ili mutacija u mehanizmu skladištenja i prenošenja genetskih informacija. Organizam čiji je gen oštećen prenijet će ga na svoje potomke na isti način kao i zdrav materijal.

U slučaju kada je patološki gen recesivan, možda se neće pojaviti u narednim generacijama, ali će oni biti njegovi nosioci. Šansa da se neće manifestirati postoji kada se pokaže da je i zdrav gen dominantan.
Trenutno je poznato više od 6 hiljada nasljednih bolesti. Mnogi od njih se pojavljuju nakon 35 godina, a neki se možda nikada neće javiti vlasniku. Dijabetes melitus, gojaznost, psorijaza, Alchajmerova bolest, šizofrenija i drugi poremećaji javljaju se izuzetno često.
Klasifikacija
Genetske bolesti koje se prenose nasljedstvom imaju ogroman broj varijeteta. Da bi se podijelili u posebne grupe, može se uzeti u obzir lokacija poremećaja, uzroci, klinička slika i priroda nasljeđa.
Bolesti se mogu klasificirati prema vrsti nasljeđivanja i lokaciji defektnog gena. Dakle, bitno je da li se gen nalazi na spolnom ili nespolnom hromozomu (autozomu), te da li je supresivan ili ne. Razlikuju se bolesti:
- Autosomno dominantna - brahidaktilija, arahnodaktilija, ectopia lentis.
- Autosomno recesivno - albinizam, mišićna distonija, distrofija.
- Ograničeno po spolu (opaženo samo kod žena ili muškaraca) - hemofilija A i B, sljepoća za boje, paraliza, fosfatni dijabetes.
Kvantitativna i kvalitativna klasifikacija nasljednih bolesti razlikuje genetske, hromozomske i mitohondrijalne tipove. Potonje se odnosi na poremećaje DNK u mitohondrijima izvan jezgra. Prva dva se javljaju u DNK, koja se nalazi u ćelijskom jezgru, i imaju nekoliko podtipova:
Monogena | Mutacije ili odsustvo gena u nuklearnoj DNK. | Marfanov sindrom, adrenogenitalni sindrom u novorođenčadi, neurofibromatoza, hemofilija A, Duchenneova miopatija. |
Poligeničan | Predispozicija i djelovanje | Psorijaza, šizofrenija, koronarna bolest, ciroza, bronhijalna astma, dijabetes melitus. |
hromozomski |
||
Promjene u strukturi hromozoma. | Miller-Dicker, Williams, Langer-Gidion sindrom. |
|
Promjena broja hromozoma. | Downov sindrom, Patauov sindrom, Edwardsov sindrom, Cleifenterov sindrom. |
|
Uzroci
Naši geni teže ne samo da akumuliraju informacije, već i da ih mijenjaju, stječući nove kvalitete. Ovo je mutacija. Javlja se prilično rijetko, otprilike 1 put u milijun slučajeva, i prenosi se na potomke ako se javlja u zametnim stanicama. Za pojedinačne gene, frekvencija mutacije je 1:108.
Mutacije su prirodni proces i čine osnovu evolucijske varijabilnosti svih živih bića. Mogu biti korisni i štetni. Neki nam pomažu da se bolje prilagodimo svom okruženju i načinu života (na primjer, suprotni palac), drugi dovode do bolesti.

Pojavu patologija u genima povećavaju fizički, hemijski i biološki faktori, neki alkaloidi, nitrati, nitriti, neki aditivi u hrani, pesticidi, rastvarači i naftni proizvodi.
Među fizičkim faktorima su jonizujuće i radioaktivno zračenje, ultraljubičasto zračenje, previsoke i niske temperature. Virusi rubeole, ospice, antigeni itd. djeluju kao biološki uzročnici.
Genetska predispozicija
Roditelji na nas utiču ne samo kroz vaspitanje. Poznato je da je kod nekih ljudi veća vjerovatnoća da će razviti određene bolesti nego kod drugih zbog naslijeđa. Genetska predispozicija za bolesti se javlja kada neko od rođaka ima abnormalnosti u genima.
Rizik od određene bolesti kod djeteta zavisi od njegovog pola, jer se neke bolesti prenose samo jednom linijom. Takođe zavisi od rase osobe i stepena odnosa sa pacijentom.
Ako osoba sa mutacijom rodi dijete, šansa da naslijedi bolest će biti 50%. Gen se možda neće ni na koji način manifestirati, jer je recesivan, a u slučaju braka sa zdravom osobom, njegove šanse da se prenesu na potomke bit će već 25%. Međutim, ako i supružnik ima takav recesivni gen, šanse za njegovo ispoljavanje kod potomaka ponovo će se povećati na 50%.
Kako prepoznati bolest?
Genetski centar će pomoći da se bolest ili predispozicija za nju otkrije na vrijeme. Obično postoji u svim većim gradovima. Prije uzimanja testova, održava se konsultacija s liječnikom kako bi se utvrdilo koji se zdravstveni problemi uočavaju kod rođaka.
Medicinsko-genetski pregled obavlja se uzimanjem krvi za analizu. Uzorak se pažljivo ispituje u laboratoriji na bilo kakve abnormalnosti. Budući roditelji obično idu na takve konsultacije nakon trudnoće. Ipak, vredi doći u genetski centar tokom njegovog planiranja.

Nasljedne bolesti ozbiljno utiču na psihičko i fizičko zdravlje djeteta i utiču na očekivani životni vijek. Većina ih je teško liječiti, a njihova se manifestacija može ispraviti samo medicinskim sredstvima. Stoga je bolje pripremiti se za to čak i prije začeća bebe.
Downov sindrom
Jedna od najčešćih genetskih bolesti je Downov sindrom. Javlja se u 13 slučajeva od 10.000 Ovo je anomalija u kojoj osoba nema 46, već 47 hromozoma. Sindrom se može dijagnosticirati odmah po rođenju.
Glavni simptomi su spljošteno lice, podignuti kutovi očiju, kratak vrat i nedostatak mišićnog tonusa. Uši su obično male, oči su koso, a oblik lubanje je nepravilan.

Bolesna djeca imaju prateće poremećaje i bolesti - upalu pluća, ARVI itd. Mogu se javiti egzacerbacije, na primjer, gubitak sluha, vida, hipotireoza, srčana oboljenja. Sa daunizmom se usporava i često ostaje na nivou od sedam godina.
Stalni rad, posebne vježbe i lijekovi značajno poboljšavaju situaciju. Mnogo je slučajeva kada su osobe sa sličnim sindromom bile sasvim sposobne da vode samostalan život, pronađu posao i postignu profesionalni uspjeh.
Hemofilija
Rijetka nasljedna bolest koja pogađa muškarce. Javlja se jednom u 10.000 slučajeva. Hemofilija nema lijeka i javlja se kao rezultat promjene jednog gena na spolnom X hromozomu. Žene su samo nosioci bolesti.

Glavna karakteristika je odsustvo proteina koji je odgovoran za zgrušavanje krvi. U tom slučaju čak i manja ozljeda uzrokuje krvarenje koje nije lako zaustaviti. Ponekad se manifestuje tek sledećeg dana nakon povrede.
Engleska kraljica Viktorija bila je nosilac hemofilije. Bolest je prenijela na mnoge svoje potomke, uključujući carevića Alekseja, sina cara Nikolaja II. Zahvaljujući njoj, bolest se počela nazivati "kraljevskom" ili "viktorijanskom".
Angelmanov sindrom
Bolest se često naziva "sindrom sretne lutke" ili "sindrom peršuna", jer pacijenti doživljavaju česte izljeve smijeha i osmeha, kao i haotične pokrete ruku. Ovu anomaliju karakteriziraju poremećaji spavanja i mentalnog razvoja.

Sindrom se javlja jednom u 10.000 slučajeva zbog odsustva određenih gena na dugom kraku hromozoma 15. Angelmanova bolest se razvija samo ako nedostaju geni na hromozomu koji je naslijeđen od majke. Kada isti geni nedostaju na očevom hromozomu, javlja se Prader-Willi sindrom.
Bolest se ne može potpuno izliječiti, ali se simptomi mogu ublažiti. U tu svrhu izvode se fizikalne procedure i masaže. Pacijenti se ne osamostaljuju u potpunosti, ali se tokom liječenja mogu sami brinuti o sebi.
Određujući formiranje fenotipa osobe tokom njenog razvoja, naslijeđe i okruženje igraju određenu ulogu u nastanku defekta ili bolesti. Međutim, doprinos genetskih faktora i faktora okoline varira između različitih stanja. Sa ove tačke gledišta, oblici odstupanja od normalnog razvoja obično se dijele u 3 grupe: nasljedne bolesti (hromozomske i genetske), multifaktorske bolesti.
Nasljedne bolesti. U zavisnosti od stepena oštećenja genetskog materijala, razlikuju se hromozomske i genske bolesti. Razvoj ovih bolesti u potpunosti je posljedica defekta u nasljednom programu, a uloga sredine je da modificira fenotipske manifestacije bolesti. U ovu grupu spadaju hromozomske i genomske mutacije i multigenske nasljedne bolesti. Nasljedne bolesti su uvijek povezane s mutacijom, međutim, fenotipske manifestacije potonje i stupanj ozbiljnosti mogu varirati kod različitih pojedinaca. U nekim slučajevima to je zbog doze mutiranog alela, u drugim – zbog utjecaja okoline.
Multifaktorske bolesti (bolesti s nasljednom predispozicijom). To su uglavnom bolesti zrele i starije životne dobi. Razlozi njihovog razvoja su faktori sredine, ali stepen njihove implementacije zavisi od genetske konstitucije organizma.
Ljudske hromozomske bolesti
Ova grupa bolesti je uzrokovana promjenama u strukturi pojedinih hromozoma ili određenog broja njih u kariotipu (neravnoteža nasljednog materijala). Kod ljudi su opisane genomske mutacije poliploidnog tipa, koje se javljaju kod pobačenih embriona (fetusa) i mrtvorođenih. Glavni dio hromozomskih bolesti su aneuploidije. Većina njih se odnosi na hromozome 21 i 22 i najčešće se nalaze u mozaicima (pojedinci koji imaju ćelije sa mutiranim i normalnim kariotipom). Trizomije su opisane za veliki broj autosoma i X hromozom (mogu biti prisutni u 4-5 kopija). Strukturna preuređivanja hromozoma takođe su obično neravnoteža genetskog materijala. Stupanj smanjenja vitalnosti ovisi o količini nedostajućeg ili viška nasljednog materijala i vrsti izmijenjenog hromozoma.
Kromosomske promjene koje dovode do razvojnih mana najčešće se unose u zigotu sa gametom jednog od roditelja. U tom slučaju, sve stanice novog organizma imat će abnormalni hromozomski set (za dijagnozu je dovoljno analizirati kariotip stanica bilo kojeg tkiva). Ako se hromozomske abnormalnosti jave u jednom od blastomera tokom prvih podjela zigote formirane od normalnih gameta, tada se razvija mozaični organizam. Da bi se utvrdila vjerovatnoća pojave hromozomske bolesti kod potomaka porodica koje već imaju bolesnu djecu, potrebno je utvrditi da li je ovaj poremećaj nov ili naslijeđen. Često roditelji bolesne djece imaju normalan kariotip, a bolest je rezultat mutacije jedne od gameta. U ovom slučaju, vjerovatnoća hromozomskog poremećaja je mala. U drugom slučaju, u somatskim ćelijama roditelja nalaze se hromozomske ili genomske mutacije, koje se mogu prenijeti na njihove zametne stanice. Kromosomske abnormalnosti na potomstvo prenose fenotipski normalni roditelji koji su nosioci uravnoteženih kromosomskih preuređivanja. Fenotipsku manifestaciju različitih hromozomskih i genomskih mutacija karakterišu rane i višestruke lezije različitih sistema i organa. Kromosomske bolesti karakterizira kombinacija mnogih urođenih mana. Također ih karakterizira raznolikost i varijabilnost fenotipskih manifestacija. Najspecifičnije manifestacije hromozomskih bolesti povezane su s neravnotežom u relativno malom fragmentu hromozoma. Neravnoteža u značajnoj količini hromozomskog materijala čini sliku manje specifičnom.
Specifičnost manifestacije kromosomske bolesti određena je promjenama u sadržaju određenih strukturnih gena koji kodiraju sintezu specifičnih proteina. Poluspecifične manifestacije hromozomske bolesti uglavnom su povezane s neravnotežom gena predstavljenih mnogim kopijama koje kontroliraju ključne procese u životu stanice. Nespecifične manifestacije kod hromozomskih bolesti povezane su sa promenama sadržaja heterohromatina u ćelijama, što utiče na normalan tok deobe ćelije, rast i formiranje kvantitativnih karakteristika.
Bolesti ljudskih gena
Među genskim bolestima razlikuju se multigenske bolesti, koje se nasljeđuju po Mendelovim zakonima, i poligenske bolesti (bolesti s nasljednom predispozicijom). U zavisnosti od funkcionalnog značaja primarnih produkata odgovarajućih gena, genske bolesti se dele na:
- kršenje enzimskog sistema (enzimopatije);
- defekti proteina u krvi (hemoglobinopatije);
- defekti u strukturnim proteinima (bolesti kolagena);
- bolesti sa nepoznatim biohemijskim defektom.
Enzimopatije
Enzimopatije se temelje ili na promjeni aktivnosti enzima ili na smanjenju intenziteta njegove sinteze. Nasljedni defekti enzima klinički se manifestiraju kod homozigotnih osoba, a kod heterozigotnih osoba nedovoljna aktivnost enzima otkriva se posebnim studijama.
U zavisnosti od metaboličkog poremećaja, razlikuju se:
- nasljedni defekti u metabolizmu ugljikohidrata (galaktozemija - poremećaj metabolizma mliječnog šećera; mukopolisaharidoza - poremećaj razgradnje polisaharida);
- nasljedni defekti u metabolizmu lipida i lipoproteina;
- nasljedni defekti u metabolizmu aminokiselina (albinizam - poremećaj sinteze pigmenta melanina; tirozinoza - poremećaj metabolizma tirozina);
- nasljedni defekti u metabolizmu vitamina;
- nasljedni defekti u metabolizmu purinskih pirimidin dušičnih baza;
- nasljedni defekti u biosintezi hormona;
- nasljedni defekti enzima crvenih krvnih zrnaca.
Hemoglobinopatije
Ova grupa bolesti je uzrokovana primarnim defektom u peptidnim lancima hemoglobina i povezanim poremećajem njegovih svojstava i funkcija (eritrocitoza, methemoglobinemija).
Kolagen ljudske bolesti
Pojava ovih bolesti zasniva se na genetskim defektima u biosintezi i razgradnji kolagena, najvažnije komponente vezivnog tkiva.
Nasljedne ljudske bolesti sa nepoznatim biohemijskim defektom
Ogroman broj monogenih nasljednih bolesti pripada ovoj grupi. Najčešći su:
- cistična fibroza - nasljedno oštećenje egzokrinih žlijezda i žljezdanih stanica tijela;
- ahondroplazija - u većini slučajeva uzrokovana novonastalom mutacijom. Ovo je bolest inertnog sistema, u kojoj se uočavaju abnormalnosti u razvoju hrskavičnog tkiva.
- mišićne distrofije (miopatije) su bolesti povezane s oštećenjem prugasto-prugastih i glatkih mišića.
Ljudske bolesti sa nasljednom predispozicijom
Ova grupa bolesti razlikuje se od genetskih bolesti po tome što zahtijevaju djelovanje faktora okoline da bi se manifestirali. Među njima su i monogene i poligene. Takve bolesti nazivaju se multifaktorskim.
Monogene bolesti sa naslednom predispozicijom su relativno malobrojne. Za njih je primjenjiva metoda Mendelejevske genetske analize. S obzirom na značajnu ulogu sredine u njihovom ispoljavanju, smatraju se nasledno determinisanim patološkim reakcijama na delovanje različitih spoljašnjih faktora (lekova, aditiva u hrani, fizičkih i bioloških agenasa), koje su zasnovane na naslednom nedostatku određenih enzima. Postavljanje tačne dijagnoze različitim metodama genetskog istraživanja, rasvjetljavanje uloge sredine i naslijeđa u nastanku bolesti, te određivanje vrste nasljeđivanja omogućavaju doktoru da razvije metode liječenja i prevencije pojave ovih bolesti u narednim generacijama.
Prilikom proučavanja prirode nasljeđivanja različitih osobina kod ljudi opisani su svi poznati tipovi nasljeđivanja i svi tipovi dominacije. Mnoge osobine se nasljeđuju monogeno, tj. određene su jednim genom i nasljeđuju se u skladu sa Mendelovim zakonima. Opisano je više od hiljadu monogenih osobina. Među njima ima i autozomnih i spolno vezanih. Neki od njih su dati u nastavku.
Monogene bolesti se javljaju kod 1-2% svjetske populacije. To je mnogo. Učestalost sporadičnih monogenih bolesti odražava učestalost procesa spontanih mutacija. Među njima, veliki udio su bolesti s biohemijskim defektom. Tipičan primjer je fenilketonurija.
Porodična manifestacija
Morphanov sindrom
Ovo je teška nasljedna bolest uzrokovana mutacijom jednog gena koja remeti normalan ciklus konverzije fenilalanina. Kod pacijenata se ova aminokiselina akumulira u stanicama. Bolest je praćena teškim neurološkim simptomima (pojačana ekscitabilnost), mikrocefalijom (mala glava) i na kraju dovodi do idiotizma. Dijagnoza se postavlja biohemijski. Trenutno, porodilišta vrše 100% skrining novorođenčadi na fenilketonuriju. Bolest je izlječiva ako se dijete odmah prebaci na posebnu ishranu koja isključuje fenilalanin.
Još jedan primjer monogene bolesti je Morphanov sindrom ili bolest paukovih prstiju. Dominantna mutacija jednog gena ima snažan pleiotropni efekat. Osim pojačanog rasta udova (prstija), kod pacijenata se javlja astenija, srčana oboljenja, iščašenje očnog sočiva i druge anomalije. Bolest se javlja u pozadini povećane inteligencije, zbog čega se naziva "bolešću velikih ljudi". Posebno su od toga patili američki predsjednik A. Lincoln i izvanredni violinista N. Paganini.
Mnoge nasljedne bolesti povezane su s promjenama u strukturi hromozoma ili njihovom normalnom broju, tj. sa hromozomskim ili genomskim mutacijama. Dakle, teška nasljedna bolest novorođenčadi, poznata kao “ sindrom mačke plakanja“, uzrokovano gubitkom (delecijom) dugog kraka hromozoma 5. Ova mutacija dovodi do abnormalnog razvoja larinksa, što uzrokuje karakterističan plač bebe. Bolest je nespojiva sa životom.

Nadaleko poznat Daunova bolest je rezultat prisustva u kariotipu dodatnog hromozoma iz 21. para (trisomija 21. hromozoma). Razlog je neraspadanje polnih hromozoma tokom formiranja zametnih ćelija kod majke. U većini slučajeva viška hromozoma kod novorođenčadi, majka navrši najmanje 35 godina života. Praćenjem učestalosti ove bolesti u područjima sa velikim zagađenjem životne sredine otkriveno je značajno povećanje broja obolelih od ovog sindroma. Takođe se pretpostavlja da će virusna infekcija uticati na majčin organizam tokom sazrevanja jajne ćelije.
Posebnu kategoriju nasljednih bolesti čine sindromi povezani s promjenama normalnog broja polnih hromozoma. Poput Daunove bolesti, javljaju se kada je poremećen proces segregacije hromozoma tokom gametogeneze kod majke.
Kod ljudi, za razliku od drozofile i drugih životinja, Y kromosom igra veliku ulogu u određivanju i razvoju spola. Ako je odsutan u skupu sa bilo kojim brojem X hromozoma, jedinka će biti fenotipski žensko, a njeno prisustvo određuje razvoj prema muškom polu. Posebno su bolesni muškarci sa hromozomskim setom XXY + 44A Klinefelterov sindrom. Karakteriziraju ih mentalna retardacija, nesrazmjeran rast udova, vrlo mali testisi, nedostatak sperme, abnormalni razvoj mliječnih žlijezda i drugi patološki znaci. Povećanje broja X hromozoma u kombinaciji sa jednim Y hromozomom ne menja definiciju muškog pola, već samo povećava Klinefelterov sindrom. XXYY kariotip je prvi put opisan 1962. godine kod 15-godišnjeg dječaka sa značajnom mentalnom retardacijom, evnuhoidnim proporcijama tijela, smanjenim testisima i rastom kose ženskog tipa. Slični znakovi su karakteristični za pacijente sa kariotipom XXXYY.
 Klinefelterov sindrom (1) i sindrom Turner-Shereshevsky (2)
Klinefelterov sindrom (1) i sindrom Turner-Shereshevsky (2)
Odsustvo jednog od dva X hromozoma u kariotipu žene (XO) uzrokuje razvoj Sindrom Turner-Shereshevsky. Obolele žene su obično niske, manje od 140 cm, zdepaste, sa slabo razvijenim mlečnim žlezdama i karakterističnim krilastim naborima na vratu. U pravilu su neplodni zbog nerazvijenosti reproduktivnog sistema. Najčešće, trudnoća s ovim sindromom dovodi do spontanog pobačaja. Samo oko 2% bolesnih žena ostane trudno do kraja.
Trisomija (XXX) ili polisomija na X kromosomu kod žena često uzrokuje bolest sličnu Turner-Shereshevsky sindromu.
Nasljedne bolesti povezane s promjenom broja X kromosoma dijagnosticiraju se citološkom metodom prema broju Barrovih tijela ili spolnog hromatina u stanicama. Godine 1949. M. Barr i C. Bertram, proučavajući interfazna jezgra neurona kod mačke, otkrili su u njima tijelo intenzivnog boja. Bio je prisutan samo u ćelijskim jezgrima ženki. Ispostavilo se da se javlja kod mnogih životinja i da je uvijek povezan sa spolom. Ova struktura se zove polni hromatin, ili Barrova tijela. Pažljivom citološkom i citogenetskom analizom ustanovljeno je da je spolni hromatin jedan od dva ženska polna hromozoma, koji je u stanju jake spiralizacije i stoga je neaktivan. Žene sa Turner-Shereshevsky sindromom (XO kariotip) ne pokazuju polni hromatin, kao i normalni XY muškarci. Normalne XX žene i abnormalni muškarci imaju po jedno Barrovo tijelo, a XXX žene i XXXY muškarci imaju dva, itd.
Osobe s nasljednim bolestima obično se rađaju sa značajnim fizičkim abnormalnostima, što omogućava da se bolest rano dijagnostikuje. Ali ponekad se bolest ne osjeti mjesecima ili čak decenijama. Na primjer, teška nasljedna bolest uzrokovana oštećenjem centralnog nervnog sistema - Huntingtonova koreja- može se pojaviti tek nakon 40 godina, a tada njegov nosilac ima vremena da ostavi potomstvo. Bolesnike karakteriziraju nevoljni trzajevi pokreti glave i udova.
Dešava se da osoba ostavlja dojam apsolutno zdrave osobe, ali ima nasljednu predispoziciju za određenu bolest, koja se manifestira pod utjecajem vanjskih ili unutrašnjih faktora. Na primjer, neki ljudi imaju tešku reakciju na određene lijekove, što je uzrokovano genetskim defektom – odsustvom određenog enzima u tijelu. Ponekad postoji fatalna reakcija na anesteziju kod naizgled zdravih ljudi, ali u stvari oni nose latentnu posebnu nasljednu bolest mišića. Kod takvih pacijenata, tokom ili nakon operacije u anesteziji, temperatura naglo raste (do 42°).
Spisak genetskih bolesti * Glavni članci: nasljedne bolesti, Nasljedne metaboličke bolesti, Enzimopatije. * U većini slučajeva daje se i šifra koja označava tip mutacije i hromozome koji su s njom povezani. također... ... Wikipedia
Ispod je lista simboličkih traka (simbolička, ili traka za obaveštenja, od engleske trake za svesnost) mali komad trake presavijen u petlju; koristi se za demonstriranje stava nosača trake prema bilo kojem pitanju ili... ... Wikipediji
Ova stranica je pojmovnik. Vidi također: Spisak genetskih malformacija i bolesti Genetski pojmovi po abecednom redu... Wikipedia
Servisna lista članaka stvorena za koordinaciju rada na razvoju teme. Ovo upozorenje nije postavljeno... Wikipedia
Grana ljudske genetike posvećena proučavanju uloge naslednih faktora u ljudskoj patologiji na svim glavnim nivoima organizacije života od populacije do molekularne genetike. Glavni dio M.g. sastoji se od kliničke genetike,...... Medicinska enciklopedija
Nasljedne bolesti su bolesti čiji je nastanak i razvoj povezan s defektima u programskom aparatu stanica, naslijeđenim putem gameta. Termin se koristi u odnosu na polietiološke bolesti, za razliku od ... Wikipedije
Bolesti, čija je pojava i razvoj povezana s defektima u programskom aparatu ćelija, naslijeđenim putem gameta. Termin se koristi u odnosu na polietiološke bolesti, za razliku od uže grupe Genetski... ... Wikipedia
Nasljedna bolest je bolest čiji je nastanak i razvoj povezan s defektima u programskom aparatu stanica, naslijeđenim putem gameta. Termin se koristi u odnosu na polietiološke bolesti, za razliku od ... ... Wikipedije
Nasljedni metabolički poremećaji uključuju veliku grupu nasljednih bolesti koje utiču na metaboličke poremećaje. Ovakvi poremećaji čine značajan dio grupe metaboličkih poremećaja (metaboličkih bolesti).... ... Wikipedia
Knjige
- Dječje bolesti, Belopolsky Yuri Arkadevič. Zdravlje djeteta bilo koje dobi je poseban zadatak za ljekara, jer organizam koji raste zahtijeva više pažnje i budnost u odnosu na bolesti. Zakazani ljekarski pregledi, identifikacija...
- Uvod u molekularnu dijagnostiku i gensku terapiju nasljednih bolesti, V. N. Gorbunova, V. S. Baranov. Knjiga iznosi savremene ideje o strukturi ljudskog genoma, metodama za njegovo proučavanje, proučavanju gena čije mutacije dovode do teške nasledne patologije: smatra se...
Nasljedne ljudske bolesti su bolesti povezane s poremećajem nasljednog aparata ćelija i naslijeđuju se s roditelja na potomstvo. Glavni rezervoar genetske informacije nalazi se u nuklearnim hromozomima. Sve ćelije ljudskog tela sadrže isti broj hromozoma u svojim jezgrama. Izuzetak su spolne ćelije ili gamete - spermatozoidi i jajašca, te mali dio stanica koje se dijele direktnom diobom. Manji dio genetskih informacija sadržan je u mitohondrijskoj DNK.
Patologija genetskog aparata javlja se na hromozomskom nivou, na nivou pojedinačnog gena, a može biti povezana i sa defektom ili odsustvom više gena. Nasljedne ljudske bolesti dijele se na:
Hromozomske bolesti
Najpoznatije hromozomske bolesti su tip trisomije - dodatni treći hromozom u paru:
- Downov sindrom - trisomija 21;
- Patau sindrom - trisomija 13;
- Edwardsov sindrom je trisomija 18 parova hromozoma.
Shereshevsky-Turnerov sindrom je uzrokovan odsustvom jednog X hromozoma kod žena.
Klinefelterov sindrom je dodatni X hromozom kod muškaraca.
Ostale hromozomske bolesti povezane su sa strukturnim preuređivanjem hromozoma kada je njihov broj normalan. Na primjer, gubitak ili udvostručenje dijela hromozoma, izmjena dijelova kromosoma iz različitih parova.

Patogeneza hromozomskih bolesti nije sasvim jasna. Očigledno, mehanizam "petog kotača" se pokreće kada odsustvo ili višak hromozoma u paru ometa normalno funkcioniranje genetskog aparata u stanicama.
Bolesti gena
Uzroci nasljednih bolesti na nivou gena su oštećenje dijela DNK, što rezultira defektom jednog specifičnog gena. Najčešće su za to odgovorne mutacije gena nasljedne degenerativne bolesti ili nasljedne metaboličke bolesti kao rezultat poremećaja sinteze odgovarajućeg strukturnog proteina ili enzima proteina:
- Cistična fibroza;
- Hemofilija;
- fenilketonurija;
- albinizam;
- Anemija srpastih ćelija;
- Intolerancija na laktozu;
- Druge metaboličke bolesti.
Monogene nasljedne bolesti se nasljeđuju prema klasičnim zakonima Gregora Mendela. Postoje autosomno dominantni, autosomno recesivni i spolno vezani tipovi nasljeđivanja.

Najčešće se realizuje genski tip nasljednih bolesti.
Bolesti s nasljednom predispozicijom ili poligene bolesti
To uključuje:
- Srčana ishemija;
- Reumatoidni poliartritis;
- Rak dojke;
- Psorijaza;
- shizofrenija;
- Alergijske bolesti;
- čir na želucu…
Lista se nastavlja i nastavlja. Postoji samo mali dio bolesti koje na ovaj ili onaj način nisu povezane s nasljednom predispozicijom. Zaista, svi procesi funkcioniranja tijela određeni su sintezom različitih proteina, kako građevinskih, tako i proteinskih enzima.

Ali ako je kod monogenih nasljednih bolesti jedan gen odgovoran za sintezu odgovarajućeg proteina, onda je kod poligenskih nasljednih bolesti nekoliko različitih gena odgovorno za složeni metabolički proces. Stoga se mutacija u jednom od njih može kompenzirati i pojaviti samo pod dodatnim vanjskim nepovoljnim uvjetima. To objašnjava da djeca sa ovim bolestima ne pate uvijek od njih, i obrnuto, djeca zdravih roditelja mogu patiti od ovih bolesti. Stoga se u slučaju poligenskih nasljednih bolesti može govoriti samo o većoj ili manjoj predispoziciji.
Dijagnoza nasljednih bolesti
Metode za dijagnosticiranje nasljednih bolesti:

Međutim, treba uzeti u obzir da su mutacije gena BRCA1 i BRCA2 odgovorne za rak dojke (BC) u samo 5-10%, a njihovo prisustvo ili odsustvo samo mijenja stepen rizika od prilično rijetkog oblika BC. Proračun efikasnosti ove metode biće predstavljen u narednim publikacijama.
Liječenje nasljednih bolesti
Simptomatsko liječenje sastoji se od korekcije metaboličkih i drugih patoloških poremećaja povezanih s ovom bolešću.
Dijetalna terapija ima za cilj eliminaciju proizvoda koji sadrže supstance koje pacijenti ne apsorbuju ili ne podnose.
Genska terapija ima za cilj uvođenje u genetski aparat ljudskih ćelija, embriona ili zigota, genetskog materijala koji nadoknađuje nedostatke u mutiranim genima. Uspjeh genske terapije je još uvijek mali. Ali medicina je optimistična u pogledu razvoja metoda genetskog inženjeringa u liječenju nasljednih bolesti.